 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | katrine Galsgaard | + 1963 word(s) | 1963 | 2020-12-25 08:48:42 | | | |
| 2 | Vicky Zhou | + 1 word(s) | 1964 | 2021-01-06 09:38:21 | | | | |
| 3 | Vicky Zhou | + 1 word(s) | 1964 | 2021-01-06 09:39:24 | | |
Video Upload Options
A key criterion for the most common chronic liver disease—non-alcoholic fatty liver disease (NAFLD)—is an intrahepatic fat content above 5% in individuals who are not using steatogenic agents or having significant alcohol intake. Subjects with NAFLD have increased plasma concentrations of glucagon, and emerging evidence indicates that subjects with NAFLD may show hepatic glucagon resistance. For many years, glucagon has been thought of as the counterregulatory hormone to insulin with a primary function of increasing blood glucose concentrations and protecting against hypoglycemia. However, in recent years, glucagon has re-emerged as an important regulator of other metabolic processes including lipid and amino acid/protein metabolism.
1. Processing and Secretion of Glucagon
Glucagon is encoded by the preproglucagon gene (GCG) [1], expressed in alpha cells, enteroendocrine L-cells, and the brain [2]. In alpha cells, GCG is processed by prohormone convertase (PC) 2 yielding glucagon [3], whereas in L-cells, GCG is processed by PC1/3, yielding glucagon-like peptide (GLP)-1, GLP-2, glicentin, and oxyntomodulin [4]. However, recent evidence suggests that the processing is not as tissue-specific as previously thought, since PC1/3 may also be active in alpha cells, giving rise to pancreatic GLP-1 [5][6][7][8], and PC2 might act in L-cells to create gut-derived glucagon [9][10][11]. It is a matter of debate whether this only occurs as a consequence of metabolic alterations, such as pancreatectomy [9][10] and bariatric surgery [11], resulting in gut-derived glucagon, or glucagon receptor inhibition [12] and streptozotocin treatment [13], resulting in pancreatic-derived GLP-1. Pancreatic GLP-1 has been suggested to be part of heathy physiology by contributing to a local incretin axis [14][15][16]. In the measurement of glucagon, analytical challenges due to cross-reactivity with other GCG-derived peptides have been an issue [17][18], and using liquid chromatography–mass spectrometry, glucagon secretion was not found to be increased after bypass surgery [19]. Upon secretion, glucagon binds to its receptor, the glucagon receptor, and regulates hepatic glucose, lipid, and amino acid/protein metabolism [20].
2. The Glucagon Receptor as a Target in the Treatment of Type 2 Diabetes, Obesity, and NAFLD
In recent years, the glucagon receptor has emerged as a target in the treatment of type 2 diabetes [21][22][23][24], obesity, and non-alcoholic fatty liver disease (NAFLD) [25][26][27]. An international consensus panel recently proposed that NAFLD should be renamed to metabolic-associated fatty liver disease (MAFLD) [28]. The criteria for MAFLD are evidence of hepatic steatosis in addition to either overweight/obesity, the presence of type 2 diabetes, or evidence of metabolic dysfunction [28]. Glucagon receptor signaling increases hepatic glucose production and a majority of subjects with type 2 diabetes show increased plasma concentrations of glucagon (hyperglucagonemia) [29][30]. Hyperglucagonemia is now known to be at least partly responsible for diabetic hyperglycemia [31][32], and therefore, clinical trials using glucagon receptor antagonists (GRAs) have been conducted in subjects with type 2 diabetes. GRA treatment improves diabetic hyperglycemia [21][22][23][24], but disturbing adverse effects including increased plasma concentrations of low-density lipoprotein and liver enzymes (aspartate- and alanine-transaminase) and hepatic fat accumulation halted further development of GRAs [33][34]. However, based on the observed changes in lipid metabolism and the possibility that glucagon may decrease food intake [35][36] and increase energy expenditure [37][38][39][40] (possibly through the sympathetic nervous system [41] or other indirect effects [42]), the focus has now shifted from glucagon receptor inhibition to glucagon receptor activation.
Glucagon analogs and agonists are being investigated as potential agents in NAFLD treatment partly due to glucagon’s regulatory effects on hepatic lipid metabolism, where glucagon increases hepatic lipolysis and fatty acid oxidation [43][44] while decreasing lipogenesis [45] and the secretion of triglycerides and very-low-density lipoprotein [46][47][48]. Consistent with this, subjects with endogenous glucagon deficiency (pancreatectomized subjects) [49] and diabetic (db/db) mice treated with glucagon receptor antisense oligonucleotide [50] have increased hepatic fat. Furthermore, glucagon has a lipid-mobilizing effect and decreases plasma concentrations of cholesterol [51][52][53][54][55], non-esterified fatty acids [44], triglycerides [52][56][57], and very-low-density lipoprotein [47], suggesting that glucagon may improve dyslipidemia.
To counteract the hyperglycemic effect of glucagon, glucagon receptor agonists are being combined with agonists of either one or both of the receptors for the incretin hormones: GLP-1 and glucose-dependent insulinotropic polypeptide [58][59][60]. Glucagon/GLP-1 co-agonists reversed obesity in diet-induced obese mice, decreased body weight to a larger degree than GLP-1 agonist comparators [61], and appear to increase energy expenditure [62]. Glucagon/GLP-1 co-agonists also improve NAFLD [63] and show promising results in models of non-alcoholic steatohepatitis, a subtype of NAFLD which potentially can progress to liver fibrosis, cirrhosis, and hepatocellular carcinoma [64], by decreasing inflammation and fibrosis while increasing mitochondrial β-oxidation, thus reducing hepatic steatosis [65]. Improvement of NAFLD has also been observed in obese or overweight individuals with type 2 diabetes treated with a glucagon/GLP-1 co-agonist [66]. Glucagon’s effect on energy expenditure and satiety may be translatable to humans, since glucagon/GLP-1 and glucagon infusions, but not the GLP-1 infusion, increased energy expenditure to a similar degree in overweight subjects without diabetes [67], and a glucagon/GLP-1 infusion decreased food intake [68]. In another human study, glucagon infusion decreased food intake but had no effect on energy expenditure, possibly due the low dose used compared to other studies [69]. The effect on energy expenditure may thus be mediated by indirect effects, possibly by glucagon acting on the GLP-1 receptor, which can be activated by high concentrations of glucagon [70][71], whereas the hepatic effects of the co-agonists may be ascribed to direct glucagon receptor activation since it, unlike the GLP-1 receptor [72], is expressed in the liver.
3. The Liver–Alpha Cell Axis May be Impaired in NAFLD
The rationale for the use of GRAs and glucagon agonists in the treatment of patients with type 2 diabetes (55–68% of whom have liver steatosis [73]) and obesity is based on glucagon’s effects on hepatic glucose and lipid metabolism, respectively. However, glucagon is also a key regulator of amino acid/protein metabolism [74][75][76][77][78], and glucagon has been suggested to be even more important for amino acid/protein metabolism than glucose metabolism [79]. Glucagon is part of an endocrine feedback loop (the liver–alpha cell axis), in which amino acids control glucagon secretion and the proliferation of alpha cells, while glucagon in turn controls hepatic amino acid metabolism and ureagenesis [80][81][82][83][84][85].
During conditions of disrupted glucagon signaling, glucagon’s effect on amino acid metabolism is impaired and as a consequence, hyperaminoacidemia develops, which in turn results in hyperglucagonemia [80][82][83][84][86][87]. In line with this, the hyperglucagonemia observed in most subjects with type 2 diabetes is associated with hyperaminoacidemia. Especially, elevated plasma levels of alanine seem to mark a disturbed liver–alpha cell axis [85][88][89][90], and alanine also appears to be a potent glucagonotropic amino acid [91]. Importantly, hyperglucagonemia seems to be associated with a fatty liver rather than with diabetes per se [92], and plasma concentrations of glucagon and non-branched-chain amino acids are characteristically increased in subjects with increased liver fat (as reflected by elevated HOMA-IR levels) [85]. Furthermore, ureagenesis is impaired in subjects with NAFLD and non-alcoholic steatohepatitis [93][94] as well as in mice with impaired glucagon receptor signaling and obese Zucker rats with hepatic steatosis [89]. Impaired liver function thus mimics the conditions of experimental glucagon deficiency (e.g., brought about by GRAs [87]), leading to the suggestion that subjects with impaired liver function due to steatosis also show glucagon resistance [95][96]. Interestingly, subjects with NAFLD appear to exhibit resistance towards glucagon-stimulated amino acid metabolism but not towards glucagon-stimulated glucose metabolism, since the hyperglycemic effect of glucagon was preserved in a pancreatic clamp study carried out in individuals with biopsy-verified NAFLD versus lean controls, whereas the effect on amino acid turnover and ureagenesis was attenuated [97]. This is consistent with the fact that the biochemical pathway of glucagon-stimulated glycogenolysis is separate from that involved in ureagenesis, whereas gluconeogenesis is closely associated with amino acid turnover and ureagenesis [98]. Considering that glucagon mainly promotes hepatic glucose production via glycogenolysis [99], this may explain why sensitivity to glucagon’s hyperglycemic effect can be preserved while its hypoaminoacidemic effect is impaired. In contrast, rats with high-fat-diet–induced hepatic steatosis showed a reduction in glucagon-stimulated hepatic glucose production, most likely due to reduced glycogenolysis [100], and this state of glucagon resistance could be reversed by exercise training, which resulted in a reduction in hepatic liver triglycerides [100]. Studies by the same group also showed that hepatic triglyceride accumulation (induced by a high-fat diet) reduced hepatic glucagon receptor density and glucagon-mediated signal transduction [101][102]. Whether this applies to human liver steatosis is unknown.
4. Hepatic Glucagon Resistance May Impair Autophagy Resulting in Increased Oxidative Stress
Exogenous glucagon stimulates hepatic autophagy [103][104][105][106], and during conditions of increased endogenous glucagon secretion, such as starvation [107], hypoglycemia [108], and type 1 diabetes [109], the number of hepatic lysosomes is increased. By stimulating autophagic protein degradation, glucagon may provide substrates for gluconeogenesis and ketogenesis and thus, sustain nutrient and energy homeostasis during starvation [110][111]. Autophagy also functions to remove damaged and non-functional organelles [112], and the autophagic degradation of mitochondria (mitophagy), which may be induced by glucagon [113], is important for maintaining mitochondrial turnover. Impaired mitophagy thus leads to the accumulation of dysfunctional mitochondria generating reactive oxygen species, resulting in oxidative stress and inflammation [114]. Autophagy is, furthermore, an important regulator of lipid metabolism and lipophagy; in other words, the degradation of triglycerides into free fatty acids in autolysosomes functions to regulate intracellular lipid stores and energy homeostasis by utilization of the released free fatty acids in mitochondrial β-oxidation [115]. Inhibition of autophagy in cultured hepatocytes and mouse livers increased triglyceride storage in lipid droplets and decreased β-oxidation and very-low-density lipoprotein secretion [116], processes which are influenced in the opposite direction by glucagon [43][44][56]. The hepatic lipolytic effect of glucagon could therefore potentially be partly mediated by lipophagy. The process of autophagy has been shown to be impaired in obesity and NAFLD [117], and hepatic resistance to glucagon in subjects with NAFLD may therefore partly explain the impaired autophagy. Amino acids, especially glutamine, leucine, and arginine, are potent activators of the mammalian target of the rapamycin complex 1 [118], which suppress autophagy [119]. The hyperaminoacidemia observed in NAFLD may therefore further impair hepatic autophagy. In NAFLD, impaired hepatic autophagy would result in the worsening/progression of NAFLD by increasing oxidative stress and lipid accumulation, and increased glucagon signaling could potentially improve NAFLD by increasing the autophagic removal of damaged organelles and lipid droplets. Whether the stimulatory effect of glucagon on hepatic autophagy is a contributing factor to the reversal of NAFLD observed by glucagon agonist treatment is currently unknown; however, the possibility provides a new perspective on the role of tri- and dual-agonists in the treatment of NAFLD.
In summary, hepatic glucagon resistance may result in a dysregulated lipid and amino acid/protein metabolism and possibly impaired autophagy, leading to excess accumulation of fat, increased oxidative stress, and hyperaminoacidemia, resulting in hyperglucagonemia and hyperglycemia. Hepatic glucagon resistance in NAFLD would thus result in the creation of a vicious circle and contribute to the worsening/progression of NAFLD (Figure 1 and Table 1). However, the majority of studies investigating these aspects of NAFLD have been performed on cells and rodents, some with contrasting findings. Further studies translating these observations to humans are thus warranted. Recognizing that glucagon’s primary role may not reside in glucose metabolism alone, but also includes important aspects of lipid and amino acid/protein metabolism, is becoming increasingly important in view of the current development of glucagon-based therapeutics for the treatment of NAFLD and obesity.
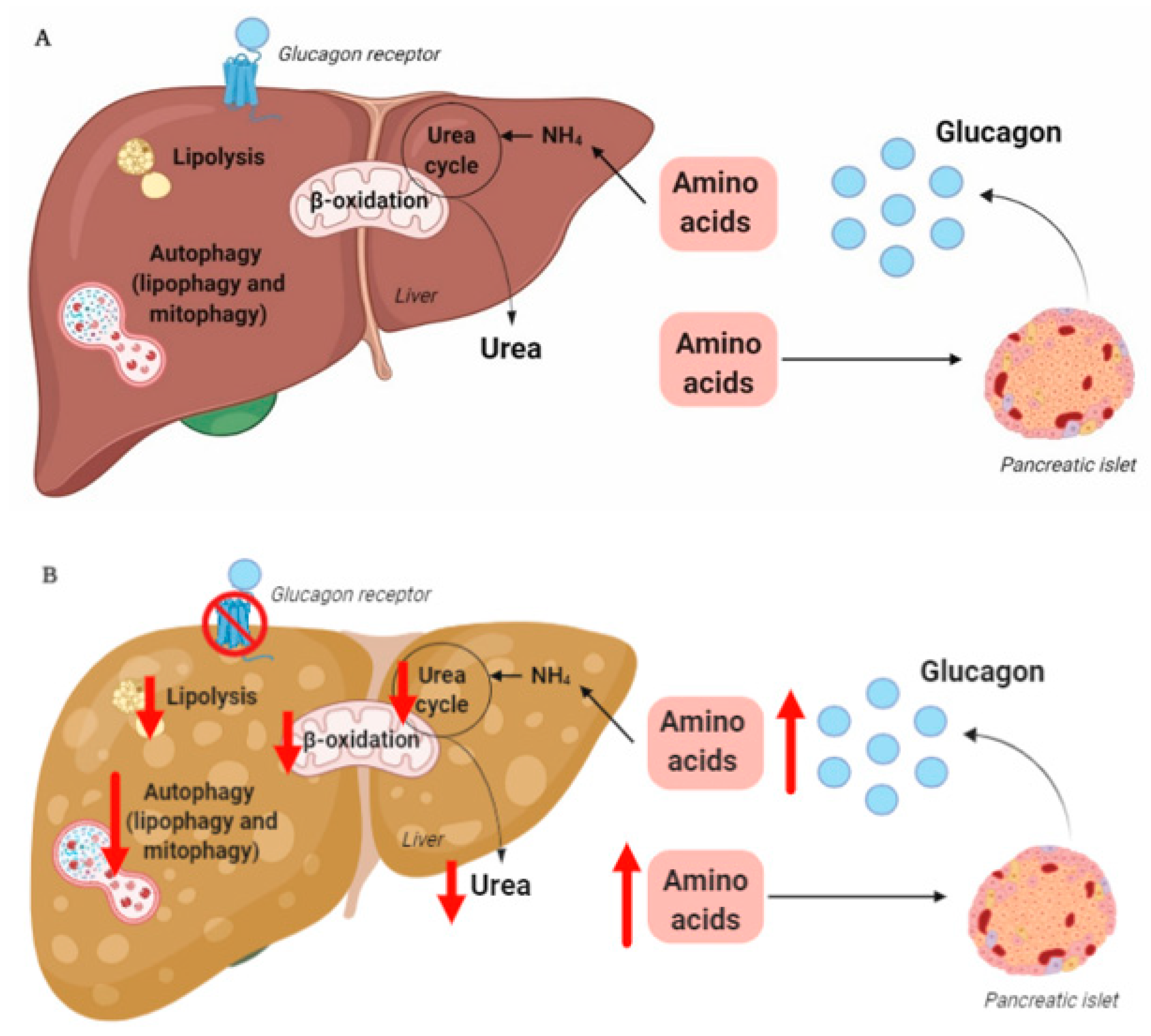
Table 1. The molecular mechanisms that may be impaired by hepatic glucagon resistance inhibition and the pathologies that are involved.
| Metabolic Process | Pathology | |
|---|---|---|
| Amino acid/protein metabolism | Amino acid transport Amino acid catabolism Ureagenesis |
Hyperaminoacidemia Hyperglucagonemia Hyperammonemia |
| Autophagy | Lipophagy Mitophagy |
Increased hepatic fat Increased oxidative stress |
| Lipid metabolism | β-oxidation Lipolysis |
Increased hepatic fat Dyslipidemia |
References
- Bell, G.I.; Sanchez-Pescador, R.; Laybourn, P.J.; Najarian, R.C. Exon duplication and divergence in the human preproglucagon gene. Nature 1983, 304, 368–371.
- Drucker, D.J. Glucagon and the Glucagon-like Peptides. Pancreas 1990, 5, 484–488.
- Rouille, Y.; Westermark, G.; Martin, S.K.; Steiner, D.F. Proglucagon is processed to glucagon by prohormone convertase PC2 in alpha TC1-6 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3242–3246.
- Rouillé, Y.; Martin, S.; Steiner, D.F. Differential Processing of Proglucagon by the Subtilisin-like Prohormone Convertases PC2 and PC3 to Generate either Glucagon or Glucagon-like Peptide. J. Biol. Chem. 1995, 270, 26488–26496.
- Kilimnik, G.; Kim, A.; Steiner, N.F.; Friedman, T.C.; Hara, M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of β-cell regeneration. Islets 2010, 2, 149–155.
- Whalley, N.M.; Pritchard, L.E.; Smith, D.M.; White, A. Processing of proglucagon to GLP-1 in pancreatic α-cells: Is this a paracrine mechanism enabling GLP-1 to act on β-cells? J. Endocrinol. 2011, 211, 99–106.
- Marchetti, P.; Lupi, R.; Bugliani, M.; Kirkpatrick, C.L.; Sebastiani, G.D.; Grieco, F.A.; Del Guerra, S.; D’Aleo, V.; Piro, S.; Marselli, L.; et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 2012, 55, 3262–3272.
- Taylor, S.W.; Nikoulina, S.E.; Andon, N.L.; Lowe, C. Peptidomic Profiling of Secreted Products from Pancreatic Islet Culture Results in a Higher Yield of Full-length Peptide Hormones than Found using Cell Lysis Procedures. J. Proteome Res. 2013, 12, 3610–3619.
- Holst, J.J.; Pedersen, J.H.; Baldissera, F.; Stadil, F. Circulating glucagon after total pancreatectomy in man. Diabetologia 1983, 25, 396–399.
- Lund, A.; Bagger, J.I.; Albrechtsen, N.J.W.; Christensen, M.; Grøndahl, M.; Hartmann, B.; Mathiesen, E.R.; Hansen, C.P.; Storkholm, J.H.; Van Hall, G.; et al. Evidence of Extrapancreatic Glucagon Secretion in Man. Diabetes 2015, 65, 585–597.
- Jorsal, T.; Albrechtsen, N.J.W.; Christensen, M.M.; Mortensen, B.; Wandall, E.; Langholz, E.; Friis, S.; Worm, D.; Ørskov, C.; Støving, R.K.; et al. Investigating Intestinal Glucagon After Roux-en-Y Gastric Bypass Surgery. J. Clin. Endocrinol. Metab. 2019, 104, 6403–6416.
- Gelling, R.W.; Du, X.Q.; Dichmann, D.S.; Romer, J.; Huang, H.; Cui, L.; Obici, S.; Tang, B.; Holst, J.J.; Fledelius, C.; et al. Lower blood glucose, hyperglucagonemia, and pancreatic cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1438–1443.
- Nie, Y.; Nakashima, M.; Brubaker, P.L.; Li, Q.-L.; Perfetti, R.; Jansen, E.; Zambre, Y.; Pipeleers, D.; Friedman, T.C. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J. Clin. Investig. 2000, 105, 955–965.
- Chambers, A.P.; Sorrell, J.E.; Haller, A.; Roelofs, K.; Hutch, C.R.; Kim, K.-S.; Gutierrez-Aguilar, R.; Li, B.; Drucker, D.J.; D’Alessio, D.A.; et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab. 2017, 25, 927–934.e3.
- Campbell, S.A.; Golec, D.P.; Hubert, M.; Johnson, J.; Salamon, N.; Barr, A.; Macdonald, P.E.; Philippaert, K.; Light, P.E. Human islets contain a subpopulation of glucagon-like peptide-1 secreting α cells that is increased in type 2 diabetes. Mol. Metab. 2020, 39, 101014.
- Fava, G.E.; Dong, E.W.; Wu, H. Intra-islet glucagon-like peptide 1. J. Diabetes Its Complicat. 2016, 30, 1651–1658.
- Albrechtsen, N.J.W.; Hartmann, B.; Veedfald, S.; Windeløv, J.A.; Plamboeck, A.; Bojsen-Møller, K.N.; Idorn, T.; Feldt-Rasmussen, B.; Knop, F.K.; Vilsbøll, T.; et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: Nonspecific interference or truly elevated levels? Diabetologia 2014, 57, 1919–1926.
- Albrechtsen, N.J.W.; Veedfald, S.; Plamboeck, A.; Deacon, C.F.; Hartmann, B.; Knop, F.K.; Vilsboll, T.; Holst, J.J. Inability of Some Commercial Assays to Measure Suppression of Glucagon Secretion. J. Diabetes Res. 2015, 2016, 1–5.
- Roberts, G.P.; Kay, R.G.; Howard, J.; Hardwick, R.H.; Reimann, F.; Gribble, F.M. Gastrectomy with Roux-en-Y reconstruction as a lean model of bariatric surgery. Surg. Obes. Relat. Dis. 2018, 14, 562–568.
- Müller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschöp, M.H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 2017, 97, 721–766.
- Kelly, R.P.; Garhyan, P.; Raddad, E.; Fu, H.; Lim, C.N.; Prince, M.J.; Pinaire, J.A.; Loh, M.T.; Deeg, M.A. Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 414–422.
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; Fu, H.; Watson, D.E.; Lewin, A.J.; Landschulz, W.H.; et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care 2015, 39, 1241–1249.
- Kazierad, D.J.; Bergman, A.; Tan, B.; Erion, D.M.; Somayaji, V.; Lee, D.S.; Rolph, T. Effects of multiple ascending doses of the glucagon receptor antagonist PF-06291874 in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2016, 18, 795–802.
- Vajda, E.G.; Logan, D.; Lasseter, K.; Armas, D.; Plotkin, D.J.; Pipkin, J.; Li, Y.-X.; Zhou, R.; Klein, D.; Wei, X.; et al. Pharmacokinetics and pharmacodynamics of single and multiple doses of the glucagon receptor antagonist LGD-6972 in healthy subjects and subjects with type 2 diabetes mellitus. Diabetes Obes. Metab. 2016, 19, 24–32.
- Henderson, S.J.; Konkar, A.; Hornigold, D.C.; Trevaskis, J.L.; Jackson, R.; Fredin, M.F.; Jansson-Löfmark, R.; Naylor, J.; Rossi, A.; Bednarek, M.A.; et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes. Metab. 2016, 18, 1176–1190.
- Zhou, J.; Cai, X.; Huang, X.; Dai, Y.; Sun, L.; Zhang, B.; Yang, B.; Lin, H.; Huang, W.; Qian, H. A novel glucagon-like peptide-1/glucagon receptor dual agonist exhibits weight-lowering and diabetes-protective effects. Eur. J. Med. Chem. 2017, 138, 1158–1169.
- More, V.R.; Lao, J.; McLaren, D.G.; Cumiskey, A.-M.; Murphy, B.A.; Chen, Y.; Previs, S.; Stout, S.; Patel, R.; Satapati, S.; et al. Glucagon like receptor 1/ glucagon dual agonist acutely enhanced hepatic lipid clearance and suppressed de novo lipogenesis in mice. PLoS ONE 2017, 12, e0186586.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209.
- Müller, W.A.; Faloona, G.R.; Aguilar-Parada, E.; Unger, R.H. Abnormal Alpha-Cell Function in Diabetes. Response to carbohydrate and protein ingestion. N. Engl. J. Med. 1970, 283, 109–115.
- Mitrakou, A.; Kelley, D.; Veneman, T.; Jenssen, T.; Pangburn, T.; Reilly, J.; Gerich, J. Contribution of Abnormal Muscle and Liver Glucose Metabolism to Postprandial Hyperglycemia in NIDDM. Diabetes 1990, 39, 1381–1390.
- Reaven, G.M.; Chen, Y.-D.I.; Golay, A.; Swislocki, A.L.M.; Jaspan, J.B. Documentation of Hyperglucagonemia Throughout the Day in Nonobese and Obese Patients with Noninsulin-Dependent Diabetes Mellitus. J. Clin. Endocrinol. Metab. 1987, 64, 106–110.
- Shah, P.; Vella, A.; Basu, A.; Basu, R.; Schwenk, W.F.; Rizza, R.A. Lack of Suppression of Glucagon Contributes to Postprandial Hyperglycemia in Subjects with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2000, 85, 4053–4059.
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528.
- Kazierad, D.J.; Chidsey, K.; Somayaji, V.R.; Bergman, A.J.; Calle, R.A. Efficacy and safety of the glucagon receptor antagonist PF-06291874: A 12-week, randomized, dose-response study in patients with type 2 diabetes mellitus on background metformin therapy. Diabetes Obes. Metab. 2018, 20, 2608–2616.
- Geary, N. Pancreatic glucagon signals postprandial satiety. Neurosci. Biobehav. Rev. 1990, 14, 323–338.
- Geary, N.; Kissileff, H.R.; Pi-Sunyer, F.X.; Hinton, V.J. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am. J. Physiol. Integr. Comp. Physiol. 1992, 262, R975–R980.
- Davidson, I.W.F.; Salter, J.M.; Best, C.H. Calorigenic Action of Glucagon. Nat. Cell Biol. 1957, 180, 1124.
- Joel, C.D. Stimulation of metabolism of rat brown adipose tissue by addition of lipolytic hormones in vitro. J. Biol. Chem. 1966, 241, 814–821.
- Doi, K.; Kuroshima, A. Modified metabolic responsiveness to glucagon in cold-acclimated and heat-acclimated rats. Life Sci. 1982, 30, 785–791.
- Nair, K.S. Hyperglucagonemia Increases Resting Metabolic Rate In Man During Insulin Deficiency. J. Clin. Endocrinol. Metab. 1987, 64, 896–901.
- Dicker, A.; Zhao, J.; Cannon, B.; Nedergaard, J. Apparent thermogenic effect of injected glucagon is not due to a direct effect on brown fat cells. Am. J. Physiol. Content 1998, 275, R1674–R1682.
- Beaudry, J.L.; Kaur, K.D.; Varin, E.M.; Baggio, L.L.; Cao, X.; Mulvihill, E.E.; Stern, J.H.; Campbell, J.E.; Scherer, P.E.; Drucker, D.J. The brown adipose tissue glucagon receptor is functional but not essential for control of energy homeostasis in mice. Mol. Metab. 2019, 22, 37–48.
- Pegorier, J.P.; Garcia-Garcia, M.V.; Prip-Buus, C.; Duee, P.H.; Kohl, C.; Girard, J. Induction of ketogenesis and fatty acid oxidation by glucagon and cyclic AMP in cultured hepatocytes from rabbit fetuses. Evidence for a decreased sensitivity of carnitine palmitoyltransferase I to malonyl-CoA inhibition after glucagon or cyclic AMP treatment. Biochem. J. 1989, 264, 93–100.
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nat. Cell Biol. 2020, 579, 279–283.
- Prip-Buus, C.; Pegorier, J.P.; Duee, P.H.; Kohl, C.; Girard, J. Evidence that the sensitivity of carnitine palmitoyltransferase I to inhibition by malonyl-CoA is an important site of regulation of hepatic fatty acid oxidation in the fetal and newborn rabbit. Perinatal development and effects of pancreatic hormones in cultured rabbit hepatocytes. Biochem. J. 1990, 269, 409–415.
- Heimberg, M.; Weinstein, I.; Kohout, M. The effects of glucagon, dibutyryl cyclic adenosine 3′,5′-monophosphate, and concentration of free fatty acid on hepatic lipid metabolism. J. Biol. Chem. 1969, 244, 5131–5139.
- Eaton, R.P. Hypolipemic action of glucagon in experimental endogenous lipemia in the rat. J. Lipid Res. 1973, 14, 312–318.
- Longuet, C.; Sinclair, E.M.; Maida, A.; Baggio, L.L.; Maziarz, M.; Charron, M.J.; Drucker, D.J. The Glucagon Receptor Is Required for the Adaptive Metabolic Response to Fasting. Cell Metab. 2008, 8, 359–371.
- Dresler, C.M.; Fortner, J.G.; McDermott, K.; Bajorunas, D.R. Metabolic Consequences of (Regional) Total Pancreatectomy. Ann. Surg. 1991, 214, 131–140.
- Liang, Y.; Osborne, M.C.; Monia, B.P.; Bhanot, S.; Gaarde, W.A.; Reed, C.; She, P.; Jetton, T.L.; Demarest, K.T. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 2004, 53, 410–417.
- Paloyan, E.; Harper, P.V. Glucagon as a regulating factor of plasma lipids. Metabolism 1961, 10, 315–323.
- Amatuzio, D.S.; Grande, F.; Wada, S. Effect of glucagon on the serum lipids in essential hyperlipemia and in hypercholesterolemia. Metabolism 1962, 11, 1240–1249.
- Penhos, J.C.; Wu, C.H.; Daunas, J.; Reitman, M.; Levine, R.; Levune, R. Effect of Glucagon on the Metabolism of Lipids and on Urea Formation by the Perfused Rat Liver. Diabetes 1966, 15, 740–748.
- Caren, R.; Corbo, L. Glucagon and cholesterol metabolism. Metabolism 1960, 9, 938–945.
- Aubry, F.; Marcel, Y.L.; Davignon, J. Effects of glucagon on plasma lipids in different types of primary hyperlipoproteinemia. Metabolism 1974, 23, 225–238.
- Guettet, C.; Mathe, D.; Riottot, M.; Lutton, C. Effects of chronic glucagon administration on cholesterol and bile acid metabolism. Biochim. Biophys. Acta 1988, 963, 215–223.
- Guettet, C.; Rostaqui, N.; Mathe, D.; Lécuyer, B.; Navarro, N.; Jacotot, B. Effect of chronic glucagon administration on lipoprotein composition in normally fed, fasted and cholesterol-fed rats. Lipids 1991, 26, 451–458.
- Gu, W.; Lloyd, D.J.; Chinookswong, N.; Komorowski, R.; Sivits, G.; Graham, M.; Winters, K.A.; Yan, H.; Boros, L.G.; Lindberg, R.A.; et al. Pharmacological Targeting of Glucagon and Glucagon-Like Peptide 1 Receptors Has Different Effects on Energy State and Glucose Homeostasis in Diet-Induced Obese Mice. J. Pharmacol. Exp. Ther. 2011, 338, 70–81.
- Sadry, S.A.; Drucker, D.J. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nat. Rev. Endocrinol. 2013, 9, 425–433.
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36.
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G.; et al. Glucagon-Like Peptide 1/Glucagon Receptor Dual Agonism Reverses Obesity in Mice. Diabetes 2009, 58, 2258–2266.
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757.
- Patel, V.C.; Joharapurkar, A.; Kshirsagar, S.; Sutariya, B.; Patel, M.; Patel, H.; Pandey, D.; Patel, D.; Ranvir, R.; Kadam, S.; et al. Coagonist of GLP-1 and Glucagon Receptor Ameliorates Development of Non-Alcoholic Fatty Liver Disease. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 35–43.
- Younossi, Z. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544.
- Jung, S.; Lee, J.; Kim, J.; Lee, Y.; Kim, Y.; Kang, J.; Trautmann, M.; Hompesch, M.; Kwon, S. Potent weight loss mechanism and improvement of NASH by the long-acting GLP-1/glucagon receptor dual agonist HM12525A. In Proceedings of the European Association for the Study of Diabetes, 51st Annual Meeting, Stockholm, Sweden, 14–18 September 2015.
- Robertson, D.; Hansen, L.; Ambery, P.; Esterline, R.L.; Jermutus, L.; Chang, Y.-T.; Petrone, M.; Johansson, E.; Johansson, L.; Sjöberg, F.B.; et al. 354-OR: Cotadutide (medi0382), a Dual Receptor Agonist with Glucagon-Like Peptide-1 and Glucagon Activity, Modulates Hepatic Glycogen and Fat Content. Diabetes 2020, 69, 354.
- Tan, T.; Field, B.C.; McCullough, K.A.; Troke, R.C.; Chambers, E.S.; Salem, V.; Maffe, J.G.; Baynes, K.C.; De Silva, A.; Viardot, A.; et al. Coadministration of Glucagon-Like Peptide-1 During Glucagon Infusion in Humans Results in Increased Energy Expenditure and Amelioration of Hyperglycemia. Diabetes 2013, 62, 1131–1138.
- Cegla, J.; Troke, R.C.; Jones, B.; Tharakan, G.; Kenkre, J.; McCullough, K.A.; Lim, C.T.; Parvizi, N.; Hussein, M.; Chambers, E.S.; et al. Coinfusion of Low-Dose GLP-1 and Glucagon in Man Results in a Reduction in Food Intake. Diabetes 2014, 63, 3711–3720.
- Bagger, J.I.; Holst, J.J.; Hartmann, B.; Andersen, B.; Knop, F.K.; Vilsbøll, T. Effect of Oxyntomodulin, Glucagon, GLP-1, and Combined Glucagon +GLP-1 Infusion on Food Intake, Appetite, and Resting Energy Expenditure. J. Clin. Endocrinol. Metab. 2015, 100, 4541–4552.
- Svendsen, B.; Larsen, O.; Gabe, M.B.N.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 2018, 25, 1127–1134.e2.
- Moens, K.; Flamez, D.; Schravendijk, C.V.; Ling, Z.; Pipeleers, D.; Schuit, F. Dual Glucagon Recognition by Pancreatic beta-Cells via Glucagon and Glucagon-Like Peptide 1 Receptors. Diabetes 1998, 47, 66–72.
- Panjwani, N.; Mulvihill, E.E.; Longuet, C.; Yusta, B.; Campbell, J.E.; Brown, T.J.; Streutker, C.; Holland, D.; Cao, X.; Baggio, L.L.; et al. GLP-1 Receptor Activation Indirectly Reduces Hepatic Lipid Accumulation but Does Not Attenuate Development of Atherosclerosis in Diabetic Male ApoE−/− Mice. Endocrinology 2013, 154, 127–139.
- Younossi, Z.; Golabi, P.; De Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801.
- Nair, K.S.; Halliday, D.; Matthews, D.E.; Welle, S.L. Hyperglucagonemia during insulin deficiency accelerates protein catabolism. Am. J. Physiol. 1987, 253, E208–E213.
- Couet, C.; Fukagawa, N.K.; Matthews, D.E.; Bier, D.M.; Young, V.R. Plasma amino acid kinetics during acute states of glucagon deficiency and excess in healthy adults. Am. J. Physiol. Metab. 1990, 258, E78–E85.
- Flakoll, P.; Borel, M.; Wentzel, L.; Williams, P.; Lacy, D.; Abumrad, N. The role of glucagon in the control of protein and amino acid metabolism in vivo. Metabolism 1994, 43, 1509–1516.
- Kraft, G.; Coate, K.C.; Winnick, J.J.; Dardevet, D.; Donahue, E.P.; Cherrington, A.D.; Williams, P.E.; Moore, M.C. Glucagon’s effect on liver protein metabolism in vivo. Am. J. Physiol. Metab. 2017, 313, E263–E272.
- Boden, G.; Rezvani, I.; Owen, O.E. Effects of glucagon on plasma amino acids. J. Clin. Investig. 1984, 73, 785–793.
- Dean, E.D. A Primary Role for Alpha Cells as Amino Acid Sensors. Diabetes 2019.
- Solloway, M.J.; Madjidi, A.; Gu, C.; Easthamanderson, J.; Clarke, H.J.; Kljavin, N.M.; Zavala-Solorio, J.; Kates, L.; Friedman, B.; Brauer, M.J.; et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of α-Cell Mass. Cell Rep. 2015, 12, 495–510.
- Holst, J.J.; Albrechtsen, N.J.W.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver–α-Cell Axis. Diabetes 2017, 66, 235–240.
- Kim, J.; Okamoto, H.; Huang, Z.; Anguiano, G.; Chen, S.; Liu, Q.; Cavino, K.; Xin, Y.; Na, E.; Hamid, R.; et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic α Cell Hyperplasia in Mice. Cell Metab. 2017, 25, 1348–1361.e8.
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation. Cell Metab. 2017, 25, 1362–1373.e5.
- Galsgaard, K.D.; Winther-Sørensen, M.; Ørskov, C.; Kissow, H.; Poulsen, S.S.; Vilstrup, H.; Prehn, C.; Adamski, J.; Jepsen, S.L.; Hartmann, B.; et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Metab. 2018, 314, E93–E103.
- Albrechtsen, N.J.W.; Færch, K.; Jensen, T.M.; Witte, D.R.; Pedersen, J.; Mahendran, Y.; Jonsson, A.E.; Galsgaard, K.D.; Winther-Sørensen, M.; Torekov, S.S.; et al. Evidence of a liver–alpha cell axis in humans: Hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 2018, 61, 671–680.
- Boden, G.; Master, R.W.; Rezvani, I.; Palmer, J.P.; Lobe, T.E.; Owen, O.E. Glucagon deficiency and hyperaminoacidemia after total pancreatectomy. J. Clin. Investig. 1980, 65, 706–716.
- Mu, J.; Qureshi, S.A.; Brady, E.J.; Muise, E.S.; Candelore, M.R.; Jiang, G.; Li, Z.; Wu, M.S.; Yang, X.; Dallas-Yang, Q.; et al. Anti-Diabetic Efficacy and Impact on Amino Acid Metabolism of GRA1, a Novel Small-Molecule Glucagon Receptor Antagonist. PLoS ONE 2012, 7, e49572.
- Pedersen, J.S.; Rygg, M.O.; Kristiansen, V.B.; Olsen, B.H.; Serizawa, R.R.; Holst, J.J.; Madsbad, S.; Gluud, L.L.; Bendtsen, F.; Albrechtsen, N.J.W. Nonalcoholic Fatty Liver Disease Impairs the Liver–Alpha Cell Axis Independent of Hepatic Inflammation and Fibrosis. Hepatol. Commun. 2020, 4, 1610–1623.
- Winther-Sørensen, M.; Galsgaard, K.D.; Santos, A.; Trammell, S.A.; Sulek, K.; Kuhre, R.E.; Pedersen, J.; Andersen, D.B.; Hassing, A.S.; Dall, M.; et al. Glucagon acutely regulates hepatic amino acid catabolism and the effect may be disturbed by steatosis. Mol. Metab. 2020, 101080.
- Gar, C.; Haschka, S.J.; Kern-Matschilles, S.; Rauch, B.; Sacco, V.; Prehn, C.; Adamski, J.; Seissler, J.; Albrechtsen, N.J.W.; Holst, J.J.; et al. The liver–alpha cell axis associates with liver fat and insulin resistance: A validation study in women with non-steatotic liver fat levels. Diabetologia 2020, 1–9.
- Felig, P. Amino Acid Metabolism in Man. Annu. Rev. Biochem. 1975, 44, 933–955.
- Albrechtsen, N.J.W.; Junker, A.E.; Christensen, M.; Hædersdal, S.; Wibrand, F.; Lund, A.M.; Galsgaard, K.D.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am. J. Physiol. Liver Physiol. 2018, 314, G91–G96.
- Eriksen, P.L.; Vilstrup, H.; Rigbolt, K.; Suppli, M.P.; Sorensen, M.; Heeboll, S.; Veidal, S.S.; Knop, F.K.; Thomsen, K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. 2019.
- Eriksen, P.L.; Sørensen, M.; Grønbæk, H.; Hamilton-Dutoit, S.; Vilstrup, H.; Thomsen, K.L. Non-alcoholic fatty liver disease causes dissociated changes in metabolic liver functions. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 551–560.
- Albrechtsen, N.J.W.; Pedersen, J.; Galsgaard, K.D.; Winther-Sorensen, M.; Suppli, M.P.; Janah, L.; Gromada, J.; Vilstrup, H.; Knop, F.K.; Holst, J.J. The liver-alpha cell axis and type 2 diabetes. Endocr. Rev. 2019.
- Suppli, M.P.; Lund, A.; Bagger, J.I.; Vilsbøll, T.; Knop, F.K. Involvement of steatosis-induced glucagon resistance in hyperglucagonaemia. Med Hypotheses 2016, 86, 100–103.
- Suppli, M.P.; Bagger, J.I.; Lund, A.; Demant, M.; Van Hall, G.; Strandberg, C.; Kønig, M.J.; Rigbolt, K.; Langhoff, J.L.; Albrechtsen, N.J.W.; et al. Glucagon Resistance at the Level of Amino Acid Turnover and Ureagenesis in Obese Subjects with Hepatic Steatosis. Diabetes 2018, 67, 147.
- Schutz, Y. Protein Turnover, Ureagenesis and Gluconeogenesis. Int. J. Vitam. Nutr. Res. 2011, 81, 101–107.
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13, 118–125.
- Charbonneau, A.; Couturier, K.; Gauthier, M.-S.; Lavoie, J.-M. Evidence of Hepatic Glucagon Resistance Associated with Hepatic Steatosis: Reversal Effect of Training. Int. J. Sports Med. 2005, 26, 432–441.
- Charbonneau, A.; Melancon, A.; Lavoie, C.; Lavoie, J.-M. Alterations in hepatic glucagon receptor density and in Gsα and Giα2 protein content with diet-induced hepatic steatosis: Effects of acute exercise. Am. J. Physiol. Metab. 2005, 289, E8–E14.
- Charbonneau, A.; Unson, C.G.; Lavoie, J.-M. High-fat diet-induced hepatic steatosis reduces glucagon receptor content in rat hepatocytes: Potential interaction with acute exercise. J. Physiol. 2007, 579, 255–267.
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202.
- Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733.
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16.
- Deter, R.L. Quantitative characterization of dense body, autophagic vacuole, and acid phosphatase-bearing particle populations during the early phases of glucagon-induced autophagy in rat liver. J. Cell Biol. 1971, 48, 473–489.
- Guder, W.; Hepp, K.D.; Wieland, O. The catabolic action of glucagon in rat liver. The influence of age, nutritional state and adrenal function on the effect of glucagon on lysosomal N-acetyl-beta, D-glucosaminidase. Biochim. Biophys. Acta 1970, 222, 593–605.
- Becker, F.F.; Cornwall, C.C. Phlorizin induced autophagocytosis during hepatocytic glycogenolysis. Exp. Mol. Pathol. 1971, 14, 103–109.
- Amherdt, M.; Harris, V.; Renold, A.E.; Orci, L.; Unger, R.H. Hepatic Autography in Uncontrolled Experimental Diabetes and Its Relationships to Insulin and Glucagon. J. Clin. Investig. 1974, 54, 188–193.
- Ruan, H.-B.; Ma, Y.; Torres, S.; Zhang, B.; Feriod, C.; Heck, R.M.; Qian, K.; Fu, M.; Li, X.; Nathanson, M.H.; et al. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev. 2017, 31, 1655–1665.
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 2011, 7, 727–736.
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593.
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 1–17.
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621.
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138.
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135.
- Lavallard, V.J.; Gual, P. Autophagy and Non-Alcoholic Fatty Liver Disease. BioMed Res. Int. 2014, 2014, 1–13.
- Zheng, L.; Zhang, W.; Zhou, Y.; Li, F.; Wei, H.; Peng, J. Recent Advances in Understanding Amino Acid Sensing Mechanisms that Regulate mTORC1. Int. J. Mol. Sci. 2016, 17, 1636.
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991.


