The rationale for the use of GRAs and glucagon agonists in the treatment of patients with type 2 diabetes (55–68% of whom have liver steatosis [
73]) and obesity is based on glucagon’s effects on hepatic glucose and lipid metabolism, respectively. However, glucagon is also a key regulator of amino acid/protein metabolism [
74,
75,
76,
77,
78], and glucagon has been suggested to be even more important for amino acid/protein metabolism than glucose metabolism [
79]. Glucagon is part of an endocrine feedback loop (the liver–alpha cell axis), in which amino acids control glucagon secretion and the proliferation of alpha cells, while glucagon in turn controls hepatic amino acid metabolism and ureagenesis [
80,
81,
82,
83,
84,
85].
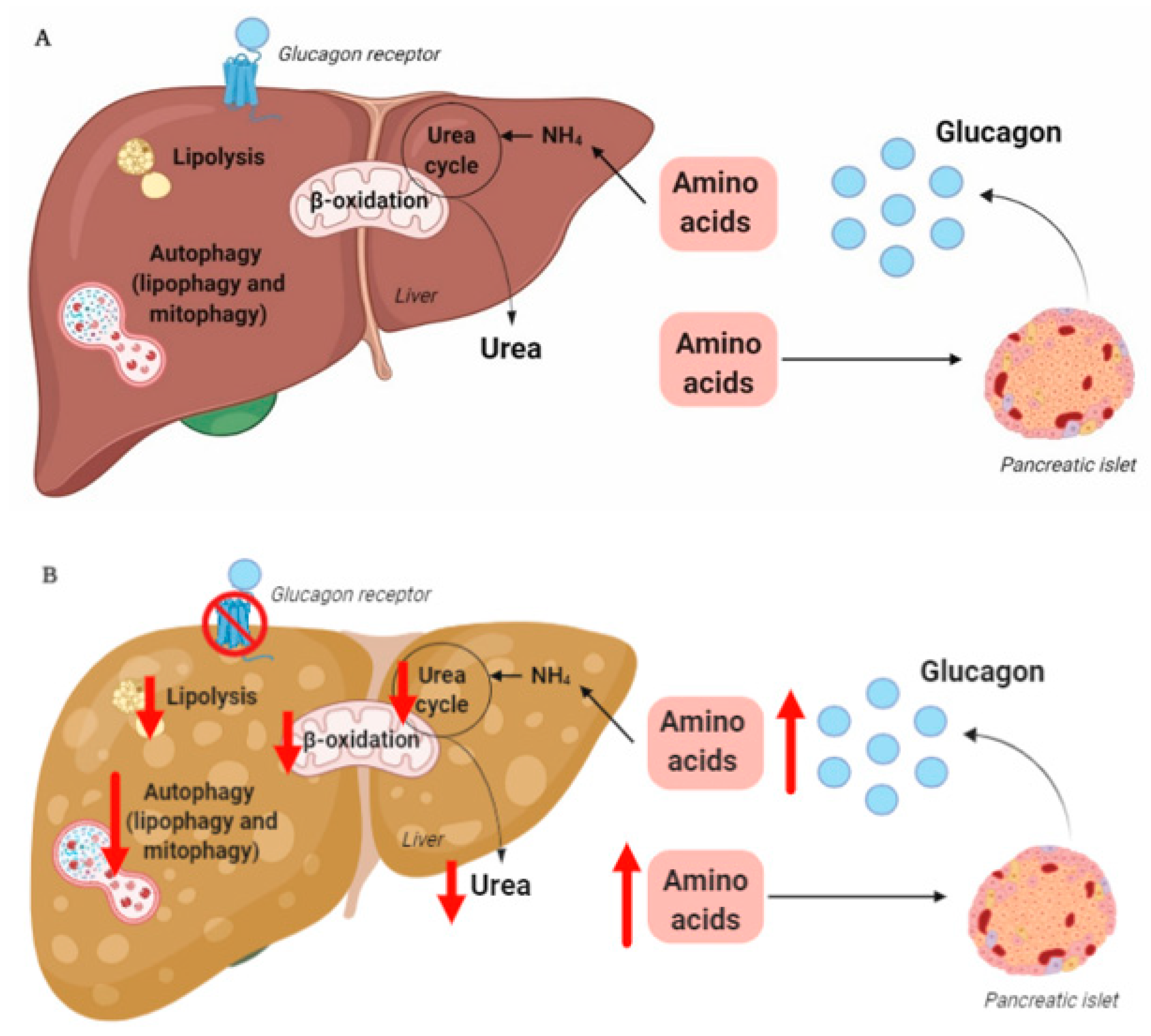
During conditions of disrupted glucagon signaling, glucagon’s effect on amino acid metabolism is impaired and as a consequence, hyperaminoacidemia develops, which in turn results in hyperglucagonemia [
80,
82,
83,
84,
86,
87]. In line with this, the hyperglucagonemia observed in most subjects with type 2 diabetes is associated with hyperaminoacidemia. Especially, elevated plasma levels of alanine seem to mark a disturbed liver–alpha cell axis [
85,
88,
89,
90], and alanine also appears to be a potent glucagonotropic amino acid [
91]. Importantly, hyperglucagonemia seems to be associated with a fatty liver rather than with diabetes per se [
92], and plasma concentrations of glucagon and non-branched-chain amino acids are characteristically increased in subjects with increased liver fat (as reflected by elevated HOMA-IR levels) [
85]. Furthermore, ureagenesis is impaired in subjects with NAFLD and non-alcoholic steatohepatitis [
93,
94] as well as in mice with impaired glucagon receptor signaling and obese Zucker rats with hepatic steatosis [
89]. Impaired liver function thus mimics the conditions of experimental glucagon deficiency (e.g., brought about by GRAs [
87]), leading to the suggestion that subjects with impaired liver function due to steatosis also show glucagon resistance [
95,
96]. Interestingly, subjects with NAFLD appear to exhibit resistance towards glucagon-stimulated amino acid metabolism but not towards glucagon-stimulated glucose metabolism, since the hyperglycemic effect of glucagon was preserved in a pancreatic clamp study carried out in individuals with biopsy-verified NAFLD versus lean controls, whereas the effect on amino acid turnover and ureagenesis was attenuated [
97]. This is consistent with the fact that the biochemical pathway of glucagon-stimulated glycogenolysis is separate from that involved in ureagenesis, whereas gluconeogenesis is closely associated with amino acid turnover and ureagenesis [
98]. Considering that glucagon mainly promotes hepatic glucose production via glycogenolysis [
99], this may explain why sensitivity to glucagon’s hyperglycemic effect can be preserved while its hypoaminoacidemic effect is impaired. In contrast, rats with high-fat-diet–induced hepatic steatosis showed a reduction in glucagon-stimulated hepatic glucose production, most likely due to reduced glycogenolysis [
100], and this state of glucagon resistance could be reversed by exercise training, which resulted in a reduction in hepatic liver triglycerides [
100]. Studies by the same group also showed that hepatic triglyceride accumulation (induced by a high-fat diet) reduced hepatic glucagon receptor density and glucagon-mediated signal transduction [
101,
102]. Whether this applies to human liver steatosis is unknown.