 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Peter Kang | + 2285 word(s) | 2285 | 2020-12-30 04:39:17 | | | |
| 2 | Vivi Li | Meta information modification | 2285 | 2020-12-31 10:44:46 | | |
Video Upload Options
Oxidative stress plays a key role in many physiological and pathological conditions. The intracellular oxidative homeostasis is tightly regulated by the reactive oxygen species production and the intracellular defense mechanisms. Increased oxidative stress could alter lipid, DNA, and protein, resulting in cellular inflammation and programmed cell death. Evidences show that oxidative stress plays an important role in the progression of various cardiovascular diseases, such as atherosclerosis, heart failure, cardiac arrhythmia, and ischemia-reperfusion injury. There are a number of therapeutic options to treat oxidative stress-associated cardiovascular diseases. Well known antioxidants, such as nutritional supplements, as well as more novel antioxidants have been studied. In addition, novel therapeutic strategies using miRNA and nanomedicine are also being developed to treat various cardiovascular diseases.
1. Introduction
Mounting evidences show that oxidative stress has an irreplaceable role in the development and pathology of various diseases [1][2][3]. It is caused by the overproduction of reactive oxygen species (ROS), which include both the free radicals and their non-radical intermediates, such as superoxide anion (O2•−), hydroxyl ion (OH•), hydrogen peroxide (H2O2), and peroxyl radicals (ROO•), alkoxyl (RO•), singlet oxygen (1O2), and ozone (O3) [4]. The burst of ROS is associated with an imbalance between the generated ROS and the antioxidant defense systems. Overproduction of ROS has a detrimental role in biological system by not only targeting biological molecules, such as lipid, protein, and DNA, but also by acting as a second messenger in cellular signaling. Through targeting regulatory pathways, ROS results in cell inflammatory signals activation or programmed cell death.
Cardiovascular diseases are the leading cause of morbidity and mortality worldwide. Evidences show that oxidative stress plays an important role in the progression of various cardiovascular diseases, such as atherosclerosis, heart failure (HF), cardiac arrhythmia, and myocardial ischemia-reperfusion (I/R) injury [5][6]. A lot of work has been devoted to the studies of antioxidants therapies in prevention and treatment of these cardiovascular disease. Small molecules, such as astaxanthin and omega-3, have shown to have a beneficial role in cardiovascular diseases. While some clinical trials have shown positive results, others are controversial. The impaired function of ROS-clearance enzymes, such as superoxide dismutase (SOD), leads to high baseline levels of oxidative stress [7]. Moreover, there are new antioxidants that are being explored, and novel strategies to specifically deliver antioxidant drugs to the area of ROS overproduction [8]. In this review, we will discuss the mechanisms of oxidative stress and their therapeutic implications in cardiovascular diseases.
2. Oxidative Stress and Cardiovascular Disease
The relationship between oxidative stress and cardiovascular diseases is shown in Figure 2.
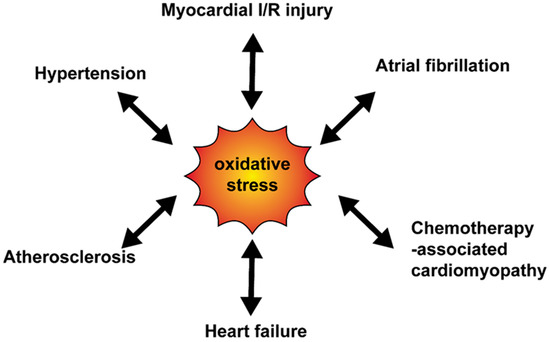
Figure 2. The relationship between oxidative stress and cardiovascular diseases. Various cardiovascular diseases enhance the oxidative production and at the same time, oxidative stress mediates progress of diseases. I/R, ischemia-reperfusion.
2.1. Oxidative Stress and Myocardial Ischemia-Reperfusion (I/R) Injury
Myocardial I/R injury is characterized by restoration of blood flow to the oxygen-deprived organs. The rapid re-establishment of blood flow leads to the oxygen burst and the ROS overproduction [9]. It is one of the most important pathogenic mechanism in acute coronary syndrome, myocardial infarction, surgical coronary bypass surgery, coronary revascularization intervention, circulatory shock, or organ transplantation [10]. Myocardial I/R injury accounts for approximately 25% of cell deaths during myocardium infarction [11]. The reperfusion injury can cause the “no-reflow” phenomenon, myocardial stunning, reperfusion arrhythmias, and reperfusion injury [12].
During reperfusion, the sources of ROS overproduction are mitochondria [13], NOX family [14], Xo [15], and uncoupled NOS [16]. Complex I and III are the major sites for ROS overproduction in myocardial I/R injury. In addition, mPTP participates in the regulation of ROS overproduction in mitochondria. Braunersreuther et al. showed that the infarcted myocardium was reduced in the NOX1 and the NOX2 deficient mice compared with the wild-type mice, while NOX4 deficient mice had no obvious phenotype [17]. At the same time, redox signaling, including the hypoxia-inducible factor (HIF) pathway [18] and Nuclear factor E2-associated factor 2 (Nrf2) [19] pathway, is also activated to antagonize the ROS burst. HIF is an oxygen sensitive transcription factor which is also regulated by NOX-related ROS production. HIF-1α attenuates I/R injury through the regulation of inducible NOS, heme oyxgenase-1, cyclooxygenase-2, and antioxidant enzymes. Studies by Li et al. showed that HIF also directly targets mitochondria and have a protective role [20]. Nrf2 is a family of transcription factors. It is located in the cytosol under normal conditions. During the oxidative stress, it is translocated into the nucleus to regulate the expression of antioxidant and anti-inflammatory factors [21].
Increased ROS is associated with cardiomyocyte mitochondria damage, DNA damage, and protein degradation, which all lead to irreversible cell death [22]. Mitochondria damages are considered as the central process of oxidative stress-mediated myocardial I/R injury. Lochner et al. showed that mitochondrial depolarization resulted in mitophagy. However, the repressed mitophagy triggered the impairment of ATP production and Ca2+ overload [23]. Myocardial I/R injury is also associated with abnormal opening of mPTP, which could lead to apoptosis or necrosis. Cyclophilin D (CyPD) resides in the mitochondrial matrix working as a scaffold to control mPTP. Evidences show that S-nitrosylation modifications of CyPD is regulated by oxidative stress; CyPD knock-in mice show less I/R injury [24].
Endoplasmic reticulum stress is also regulated by oxidative stress and plays an important role in myocardial I/R injury [25]. Oxidative stress modifies amino acid residues to regulate protein activities and disturb intracellular Ca2+-homeostasis. In addition, inflammation is of great importance in myocardial I/R injury process. After myocardial I/R injury, cytokine cascades, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), are activated. The activated cytokines will induce inflammation in these cells. Among them, neutrophils are the predominant early responder [26]. Neutrophils recruited to the infarct zone contain high levels of NOX2 and MPO, which facility the production of ROS. Furthermore, cytokine cascade has been shown to suppress cardiac contractility and decrease collagen synthesis [27].
A clinical study in patients undergoing the primary percutaneous coronary intervention showed plasma 8-iso-prostaglandin F2alpha, which is used as an indicator of oxidative stress, was increased after the procedure. However, there was no relationship between 8-iso-prostaglandin F2alpha and troponin T [28]. In patients undergoing coronary artery bypass surgery, thiobarbituric acid reactive substances (TBARS) was measured to indicate the oxidative stress status. The results showed that the oxidative stress was increased after surgery and the peak increase was seen 1 h after the reperfusion [29].
2.2. Oxidative Stress and Heart Failure (HF)
HF is characterized by the inadequate cardiac output to meet the bodily demands. Clinically, it is manifested by shortness of breath and/or chest tightness [30]. Studies showed that ROS are overproduced in all stages of HF [31]. Interestingly, the main source of ROS is different in HF patients with or without reduced ejection fraction. HF with reduced ejection fraction (HFrEF) is characterized by a reduced ejection fraction and is commonly due to coronary artery disease (CAD). In HFrEF, the injured cardiomyocytes produce ROS which leads to the maladaptive remodeling through programmed cell death and fibrosis [32]. In contrast, HF with preserved ejection fraction (HFpEF) is usually caused by hypertension, diabetes or genetics (e.g., hypertrophic cardiomyopathy). In HFpEF, the endothelial cells are the major sites of ROS overproduction [32][33]. The risk factors associated with HFpEF result in elevated plasma proinflammatory factors, such as IL-6, soluble ST2, and TNF-α. These factors act on the endothelial cells to induce ROS production, which upregulate the protein kinase G signaling in cardiomyocytes.
Mitochondria are major sites of ROS production in failing hearts while NOX and Xo activities are also increased [34][35]. The overproduction of ROS in HF patients causes mitochondria damage, which gives feedback to produce more ROS [36]. At the same time, ROS accelerates myocardial remodeling by activating variety of hypertrophic signaling kinases and transcription factors, such as tyrosine kinase Src, GTP-binding protein Ras, and mitogen-activated protein kinases (MAPKs) [37][38]. In addition, matrix metalloproteinases, important factors for myocardial structure, are also activated by ROS [39].
It is worth noting that the chemotherapy associated cardiomyopathy is a special kind of HF. It is relatively common and serious complication of chemotherapy [40]. Many classes of chemotherapeutic agents that are widely used in the clinics are identified to have cardiac toxicity, such as anthracyclines and alkylating agents [41]. These agents cause the increase in ROS generation and enhance the oxidative stress in the cell, which then leads to cardiomyocyte death [42]. Doxorubicin (DOX), a widely used anthracycline chemotherapeutic drug, directly modifies the mitochondrial DNA and disturbs the mitochondrial function, the protein expression, and the lipid peroxidation [42]. DOX combines with free iron to form iron-Dox complex, then reacts with oxygen and facilitates ROS production [43].
Clinical study by Tedgui showed that pericardial levels of 8-iso-prostaglandin F2alpha was associated with severity of HF [44]. The study by Chopra et al. investigated plasma lipid peroxides (MDA) in congestive HF patients and found an inverse correlation between MDA and left ventricular ejection fraction [45].
2.3. Oxidative Stress and Atherosclerosis
Atherosclerosis is the underlying pathology of ischemia heart diseases, stroke and peripheral artery diseases. Oxidative stress is essential for the pathological progress of atherosclerosis. Development of atherosclerotic plaques will decrease the oxygen supply which is the basis of many kinds of cardiovascular diseases [46]. Atherosclerosis is initiated by the injury of endothelial cells, followed by the infiltration and accumulation of oxidized low-density lipoprotein (ox-LDL) cholesterol to the subendothelial space. At the same time, leukocytes migrate to the subendothelial space. Monocytes-originated macrophages engulf ox-LDL to form foam cells [47]. NOX, Xo, mitochondrial enzymes are mainly responsible for the production of ROS in atherosclerosis [48]. NOX1 [49] and NOX4 [50] are detected in vascular smooth muscle cells (VSMCs). In comparison, NOX2 [51], NOX4 [52], and Xo [53] are found in endothelial cells.
Oxidative stress regulates the pathophysiology of atherosclerosis at all stages. First, oxidative stress causes endothelial dysfunction by altering endothelial signal transduction and redox-regulated transcription factors, which increase vascular endothelial permeability and catalyze leukocyte adhesion. This is considered as the initiation of plaque formation. Then, plasma LDL is recruited to the arterial wall where it is modified by oxidative stress to form ox-LDL. Ox-LDL can be taken up by macrophage to form foam cells. In addition, oxidative stress alters the expression of adhesion molecules, such as a vascular cell adhesion molecule-1, to regulate adhesion of monocytes. At the same time, increased ROS stimulate the development of the plaque by enhancing VSMCs migration and collagen deposition. Finally, oxidative stress exacerbates the stability of the plaque by releasing matrix metalloproteinase to degrade the fibrous wall [54][55].
Channon et al. showed the association between endothelial dysfunction and increased vascular superoxide production in human atherosclerosis [56]. Other study demonstrating the increase in erythrocyte TBARS with the severity of obstruction of the artery supports the potential causal relationship between oxidative stress and atherosclerosis [57].
2.4. Oxidative Stress and Atrial Fibrillation (AF)
AF is the most common arrhythmia in clinical practice with symptoms of irregular and rapid heart rate [58]. In rats, decreased plasma antioxidant capacity was associated with increased risk of AF [59]. Quyyumi et al. showed that there were elevated cystine level, cystine/glutathione ratio, and redox potential of glutathione (all indictive of increased oxidative stress) in AF patients [60]. The sources of ROS include NOX2/4 enzymes, which is upregulated in fibrillating area, and Xo [61].
There may be multiple mechanisms for how ROS causes AF. First, the increase in ROS regulates ionic leaks in cardiomyocytes. It increases Na+ current [62], L-type Ca2+ current [63], and Ca2+ leak from the sarcoplasmic reticulum (SR) [64]. All these result in prolonged action potential duration and reduced conduction velocity [65]. Furthermore, oxidative stress promotes myocardial fibrosis by facilitating the deposition of collagen [66]. The myocardial fibrosis interferes with the electrical coupling of myocytes [67]. Finally, oxidative stress may cause AF by regulating iron current associated proteins, DNA and post-translational modifications. Angiotensin II and hypoxia result in Na+ current abnormality by regulating Na+ voltage-gated channel alpha subunit 5 (SCN5A) splicing mRNA. Ca2+/CaM-dependent kinase II (CaMKII) is oxidized by ROS and has potential to regulate Na+ current by ryanodine receptor, an important Ca2+ control protein located in the SR [68][69].
2.5. Oxidative Stress and Hypertension
Hypertension is regarded as the major risk factor for cardiovascular diseases. It is a pathologic blood pressure increase resulting from the abnormal vasorelaxation factor levels. Furthermore, laboratory studies showed that oxidative stress levels in hypertension models differ from control group [70][71]. Similar to atherosclerosis, NOX families are regarded as a major source of ROS while Xo, NOS, mitochondria also have important roles in ROS increase [72][55]. Oxidative stress regulates hypertension by targeting endothelial cells. Vascular tonicity is regulated by the balance of endothelium-derived relaxing (EDRFs) factors and endothelium-derived hyperpolarizing factors (EDHFs). ROS is known as a member of EDHFs, whereas nitric oxide (NO) is a member of EDRFs [73]. ROS is able to decrease the bioavailability of NO and increase the amount of endogenous endothelial NOS antagonist, such as asymmetric dimethylarginine (ADMA). Additionally, endothelial function is regulated by cell phosphorylation pathways, such as tyrosine kinases, phosphoinositol-3-kinase/Akt kinase (PI3K/Akt) and the MAPKs, and the gene expression factors, such as p53 and activated protein-1 (AP-1). All these pathways and gene expression factors can be initiated and controlled by oxidative stress [54][74].
Chayama et al. measured urinary excretion of 8-hydroxy-2′-deoxyguanosine and serum MDA-modified LDL as the indicator of oxidative stress in patients with renovascular hypertension. They showed that there was an increase in oxidative stress indicators in these hypertensive patient compared to the control group without hypertension [75]. Another study by Beevers also showed that the lipid hydroperoxides were upregulated in hypertension patients [76].
3. Conclusions
Oxidative stress plays an important role in the development and the evolution of cardiovascular diseases. Various therapeutic strategies targeting oxidative stress have been developed. Although animal studies have shown beneficial effects of antioxidant therapy in various cardiovascular diseases, the clinical outcomes vary in human trials. The deeper understanding of oxidative stress in the cardiovascular diseases and development of better antioxidants therapies are needed to have more effective treatments of cardiovascular diseases.
References
- Bisht, S.; Faiq, M.; Tolahunase, M.; Dada, R. Oxidative stress and male infertility. Nat. Rev. Urol. 2017, 14, 470–485.
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273.
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281.
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299.
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Biesalski, H.K.; Grune, T.; Tinz, J.; Zöllner, I.; Blumberg, J.B. Reexamination of a meta-analysis of the effect of antioxidant supplementation on mortality and health in randomized trials. Nutrients 2010, 2, 929–949.
- Kim, K.S.; Song, C.G.; Kang, P.M. Targeting Oxidative Stress Using Nanoparticles as a Theranostic Strategy for Cardiovascular Diseases. Antioxid. Redox Signal. 2019, 30, 733–746.
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123.
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401.
- Binder, A.; Ali, A.; Chawla, R.; Aziz, H.A.; Abbate, A.; Jovin, I.S. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev. Cardiovasc. Ther. 2015, 13, 1045–1057.
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135.
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819.
- Kahles, T.; Brandes, R.P. Which NADPH Oxidase Isoform Is Relevant for Ischemic Stroke? The Case for Nox 2. Antioxid. Redox Signal. 2013, 18, 1400–1417.
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606.
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551.
- Braunersreuther, V.; Montecucco, F.; Asrih, M.; Ashri, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quinderé, A.L.G.; Montessuit, C.; et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64.
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24.
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647.
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109.
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502.
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89.
- Dhanabalan, K.; Mzezewa, S.; Huisamen, B.; Lochner, A. Mitochondrial Oxidative Phosphorylation Function and Mitophagy in Ischaemic/Reperfused Hearts from Control and High-Fat Diet Rats: Effects of Long-Term Melatonin Treatment. Cardiovasc. Drugs Ther. 2020.
- Amanakis, G.; Sun, J.; Fergusson, M.M.; McGinty, S.; Liu, C.; Molkentin, J.D.; Murphy, E. Cysteine 202 of Cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc. Res. 2020.
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875.
- El Kazzi, M.; Rayner, B.S.; Chami, B.; Dennis, J.M.; Thomas, S.R.; Witting, P.K. Neutrophil-Mediated Cardiac Damage after Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid. Redox Signal. 2020, 33, 689–712.
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36.
- Berg, K.; Jynge, P.; Bjerve, K.; Skarra, S.; Basu, S.; Wiseth, R. Oxidative stress and inflammatory response during and following coronary interventions for acute myocardial infarction. Free Radic. Res. 2005, 39, 629–636.
- Akila; D’Souza, B.; Vishwanath, P.; D’Souza, V. Oxidative injury and antioxidants in coronary artery bypass graft surgery: Off-pump CABG significantly reduces oxidative stress. Clin. Chim. Acta 2007, 375, 147–152.
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Compr. Physiol. 2015, 6, 187–214.
- LeLeiko, R.M.; Vaccari, C.S.; Sola, S.; Merchant, N.; Nagamia, S.H.; Thoenes, M.; Khan, B.V. Usefulness of elevations in serum choline and free F2)-isoprostane to predict 30-day cardiovascular outcomes in patients with acute coronary syndrome. Am. J. Cardiol. 2009, 104, 638–643.
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Münzel, T.; Gori, T.; Keaney, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564.
- Sawyer, D.B.; Colucci, W.S. Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ. Res. 2000, 86, 119–120.
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.-L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171.
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168.
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid. Redox Signal. 2003, 5, 731–740.
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190.
- Kameda, K.; Matsunaga, T.; Abe, N.; Hanada, H.; Ishizaka, H.; Ono, H.; Saitoh, M.; Fukui, K.; Fukuda, I.; Osanai, T.; et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease. Possible role for left ventricular remodelling. Eur. Heart J. 2003, 24, 2180–2185.
- Cai, A.W.; Taylor, M.H.; Ramu, B. Treatment of chemotherapy-associated cardiomyopathy. Curr. Opin. Cardiol. 2019, 34, 296–302.
- Yu, A.F.; Steingart, R.M.; Fuster, V. Cardiomyopathy Associated with Cancer Therapy. J. Card. Fail. 2014, 20, 841–852.
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642.
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48.
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539.
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248.
- Scicchitano, P.; Cortese, F.; Gesualdo, M.; De Palo, M.; Massari, F.; Giordano, P.; Ciccone, M.M. The role of endothelial dysfunction and oxidative stress in cerebrovascular diseases. Free Radic. Res. 2019, 53, 579–595.
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380.
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219.
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894.
- Ellmark, S.H.M.; Dusting, G.J.; Ng Tang Fui, M.; Guzzo-Pernell, N.; Drummond, G.R. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc. Res. 2005, 65, 495–504.
- Görlach, A.; Brandes, R.P.; Nguyen, K.; Amidi, M.; Dehghani, F.; Busse, R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ. Res. 2000, 87, 26–32.
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233.
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551.
- Ahmad, K.A.; Yuan Yuan, D.; Nawaz, W.; Ze, H.; Zhuo, C.X.; Talal, B.; Taleb, A.; Mais, E.; Qilong, D. Antioxidant therapy for management of oxidative stress induced hypertension. Free Radic. Res. 2017, 51, 428–438.
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471.
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, E85–E90.
- Azarsiz, E.; Kayikcioglu, M.; Payzin, S.; Yildirim Sözmen, E. PON1 activities and oxidative markers of LDL in patients with angiographically proven coronary artery disease. Int. J. Cardiol. 2003, 91, 43–51.
- Lau, D.H.; Nattel, S.; Kalman, J.M.; Sanders, P. Modifiable Risk Factors and Atrial Fibrillation. Circulation 2017, 136, 583–596.
- Kalani, R.; Judge, S.; Carter, C.; Pahor, M.; Leeuwenburgh, C. Effects of caloric restriction and exercise on age-related, chronic inflammation assessed by C-reactive protein and interleukin-6. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 211–217.
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855.
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484.
- Morita, N.; Lee, J.H.; Xie, Y.; Sovari, A.; Qu, Z.; Weiss, J.N.; Karagueuzian, H.S. Suppression of Re-Entrant and Multifocal Ventricular Fibrillation by the Late Sodium Current Blocker Ranolazine. J. Am. Coll. Cardiol. 2011, 57, 366–375.
- Thomas, G.P.; Sims, S.M.; Cook, M.A.; Karmazyn, M. Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. J. Pharmacol. Exp. Ther. 1998, 286, 1208–1214.
- Anzai, K.; Ogawa, K.; Kuniyasu, A.; Ozawa, T.; Yamamoto, H.; Nakayama, H. Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem. Biophys. Res. Commun. 1998, 249, 938–942.
- Sovari, A.A. Cellular and Molecular Mechanisms of Arrhythmia by Oxidative Stress. Cardiol. Res. Pract. 2016, 2016.
- Ishii, Y.; Schuessler, R.B.; Gaynor, S.L.; Yamada, K.; Fu, A.S.; Boineau, J.P.; Damiano, R.J. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation 2005, 111, 2881–2888.
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.F.; Chen, P.S.; Xie, L.H.; Chen, F.; et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1594–H1605.
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3.
- Ren, X.; Wang, X.; Yuan, M.; Tian, C.; Li, H.; Yang, X.; Li, X.; Li, Y.; Yang, Y.; Liu, N.; et al. Mechanisms and Treatments of Oxidative Stress in Atrial Fibrillation. Curr. Pharm. Des. 2018, 24, 3062–3071.
- Rodrigo, R.; Prat, H.; Passalacqua, W.; Araya, J.; Guichard, C.; Bächler, J.P. Relationship between Oxidative Stress and Essential Hypertension. Hypertens. Res. 2007, 30, 1159–1167.
- Touyz, R.M.; Schiffrin, E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: Role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J. Hypertens. 2001, 19, 1245–1254.
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667.
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540.
- Dinh, Q.N.; Drummond, G.R.; Sobey, C.G.; Chrissobolis, S. Roles of Inflammation, Oxidative Stress, and Vascular Dysfunction in Hypertension. BioMed Res. Int. 2014, 2014.
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962.
- Lip, G.Y.H.; Edmunds, E.; Nuttall, S.L.; Landray, M.J.; Blann, A.D.; Beevers, D.G. Oxidative stress in malignant and non-malignant phase hypertension. J. Hum. Hypertens. 2002, 16, 333–336.


