 +1 credit
+1 credit
Video Upload Options
Phosphonopeptides are phosphorus analogues of peptides and have been widely applied as enzyme inhibitors and antigens to induce catalytic antibodies. Phosphonopeptides generally contain one aminoalkylphosphonic acid residue and include phosphonopeptides with C-terminal aminoalkylphosphonic acids and phosphonopeptides with a phosphonamidate bond. The phosphonamidate bond in the phosphonopeptides is generally formed via phosphonylation with phosphonochloridates, condensation with coupling reagents and enzymes, and phosphinylation followed by oxidation. Pseudo four-component condensation reaction of amides, aldehydes, alkyl dichlorophosphites, and amino/peptide esters is an alternative, convergent, and efficient strategy for synthesis of phosphonopeptides through simultaneous construction of aminoalkylphosphonic acids and formation of the phosphonamidate bond.
1. Introduction
Phosphonopeptides are phosphorus analogues of peptides. They generally contain one aminoalkylphosphonic acid residue and include phosphonopeptides with C-terminal aminoalkylphosphonic acids and phosphonopeptides containing a phosphonamidate bond [1][2]. Phosphonopeptides have been used as antibacterial agents [3]. They have been widely applied as enzyme inhibitors [4][5][6][7][8] and antigens for inducing catalytic antibodies [9][10][11][12] due to their tetrahedral structural feature. Phosphonopeptides containing C-terminal aminoalkylphosphonic acids have been prepared via coupling of N-protected amino acyl chlorides with aminoalkylphosphonic acids [13], condensation of N-protected amino acids or peptides and aminoalkylphosphonic acids with coupling reagents [14][15], aminolysis of N-chloroacetyl aminoalkylphosphonic acids [16], and the Mannich-type reactions of N-protected amino amides or aminoalkanesulfonamides, aldehydes, and phosphorus trichloride followed by hydrolysis [17][18]. Synthesis of phosphonopeptides with C-terminal aminoalkylphosphonic acids was reviewed recently [19]. This review focuses on the synthetic methods of phosphonopeptides, including α-, β-, γ-, and δ-phosphonopeptides (Figure 1), with a phosphonamidate bond, especially focuses on the synthetic strategies for the formation of the phosphonamidate bond, excluding the modification of phosphonopeptides.
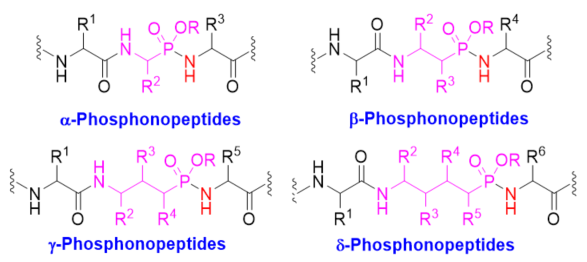
Figure 1. Different classes of phosphonopeptides.
2. Synthesis of Phosphonopeptides via Phosphonochloridates
Phosphonylation of amino/peptide esters with alkyl N-protected aminoalkylphosphonochloridates is a general and widely applied method for the synthesis of phosphonopeptides containing a phosphonamidate bond. The phosphonochloridates are usually prepared via chlorination of the corresponding dialkyl phosphonates with phosphorus pentachloride [20] or phosphorus oxychloride [21], chlorination of phosphonic monoesters with thionyl chloride [22][23] or oxalyl chloride [24][25], and chlorination of alkyl trimethylsilyl phosphonates [26] or alkyl phosphinates [27] with carbon tetrachloride. Phosphonobromidates are seldom applied as intermediates in the synthesis of phosphonopeptides and generated via bromination of alkyl phosphinates with bromine [28]. Each of the above-mentioned methods will be presented as following.
2.1. Chlorination of Dialkyl Phosphonates with Phosphorus Pentachloride
After aminomethylphosphonic acid was isolated from numerous organisms and animal and human organs [29][30], to understand the biological significance of this new class of compounds, phosphonodipeptide was synthesized in 1973. Diisopropyl N-phthalyl(Phth)aminomethylphosphonate was prepared from phthalylaminomethyl chloride and triisopropyl phosphite and further treated with phosphorus pentachloride to give the corresponding isopropyl N-phthalylaminomethylphosphonochloridate, which reacted with ethyl glycinate in the presence of triethylamine to give rise to protected phosphonopeptide (Scheme 1). This is the first chemical synthesis of α-phosphonopeptide with a phosphonamidate linkage.
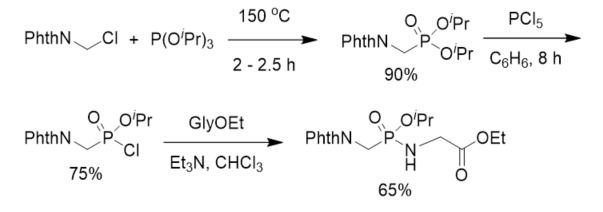
Scheme 1. First synthesis of phosphonopeptide.
Similarly, N-phthalylaminomethylphosphonochloridate was prepared and further coupled with dipeptide esters to afford phosphonotripeptides. After hydrazinolysis and acetylation with acetic anhydride and acylation with acyl chlorides or N-benzyloxycarbonyl(Cbz)-protected dipeptides, the phosphonotripeptides were transformed into N-acetyl phosphonotripeptides, N-acyl phosphonotripeptides, and phosphonopentapeptides after hydrogenolysis, respectively (Scheme 2). As inhibitors of enkephalinase and angiotensin-converting enzyme (ACE), these phosphonopeptides exhibited good inhibitory potency against enkephalinase with several of the analogs having Ki values in the submicromolar range as contrasted to micromolar or higher toward ACE [20]. Another series of phosphonopeptides were synthesized as potential inhibitors of ACE [21]. The phosphonamide bond in the phosphonopeptides is generally stable under weak acidic and basic conditions.
Scheme 2. Synthesis of phosphonopeptides as inhibitors of enkephalinase and angiotensin-converting enzyme.
2.2. Chlorination of Dialkyl Phosphonates with Phosphorus Oxychloride
Besides phosphorus pentachloride, phosphorus oxychloride was also applied in the conversion of dialkyl phosphonates into phosphonochloridates. Phosphonopeptide was synthesized from ethyl glycylglycinate hydroloride and ethyl phosphonochloridate derived from direct chlorination of diethyl phosphonate with phosphorus oxychloride (Scheme 3) [21].
Scheme 3. Synthesis of phosphonopeptide from phosphonochloridate generated by chlorination of diethyl phosphonate with phosphorus oxychloride.
2.3. Chlorination of Alkyl Phosphonic Acid Monoesters with Thionyl Chloride
To search for competitive inhibitors of d-alanine:d-alanine ligase, phosphonodipeptide was prepared from methyl alaninate and N-Cbz-protected methyl 1-aminoethylphosphonochloridate, which was prepared from N-Cbz-protected diphenyl 1-aminoethylphosphonate via transesterification, selective hydrolysis, and chlorination with thionyl chloride (Scheme 4). The phosphonodipeptide was the competitive inhibitor of d-alanine:d-alanine ligase, with Ki close to 10−6 M [22].
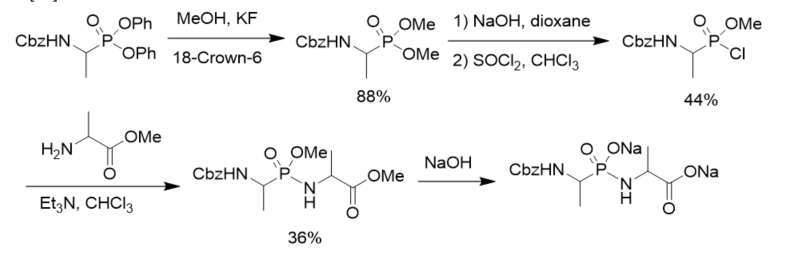
Scheme 4. Synthesis of phosphonopeptide as the inhibitor of d-alanine:d-alanine ligase.
Some α-phosphonodipeptides which simulated the transition state of d-alanyl-d-alanine synthetase (EC 6.3.2.4) reaction were synthesized (Scheme 5). The inhibition of the synthetase by these phosphonopeptides was studied with the S. faecalis enzyme. The phosphonopeptides exhibited time-dependent inhibition in the presence of ATP, suggesting that they underwent phosphorylation prior to inactivating the enzyme [23].
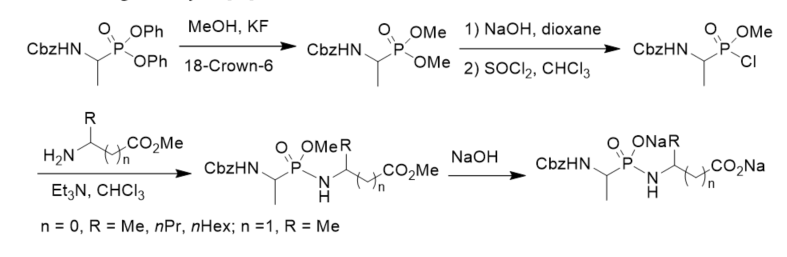
Scheme 5. Synthesis of phosphonopeptides from aminoalkylphosphintes.
N-Cbz-protected diethyl 1-aminoalkylphosphonates were prepared via three component condensation reaction of benzyl carbamate, aldehydes, and diethyl phosphite in acetyl chloride. After hydrogenolysis and coupled with N-protected amino esters, they were converted into phosphonodipeptides with C-terminal 1-aminoalkylphosphonic acids. N-Cbz-protected diethyl 1-aminoalkylphosphonates were transformed to N-Cbz-protected 1-aminoalkylphosphonic monoesters via basic hydrolysis and treatment with thionyl chloride and alcohol. N-Cbz-protected 1-aminoalkylphosphonic monoesters were treated with thionyl chloride followed by reactions with amino esters or 1-aminoalkylphosphonates, affording phosphonopeptides composing of one or two 1-aminoalkylphosphonic acid residue(s) (Scheme 6) [31]
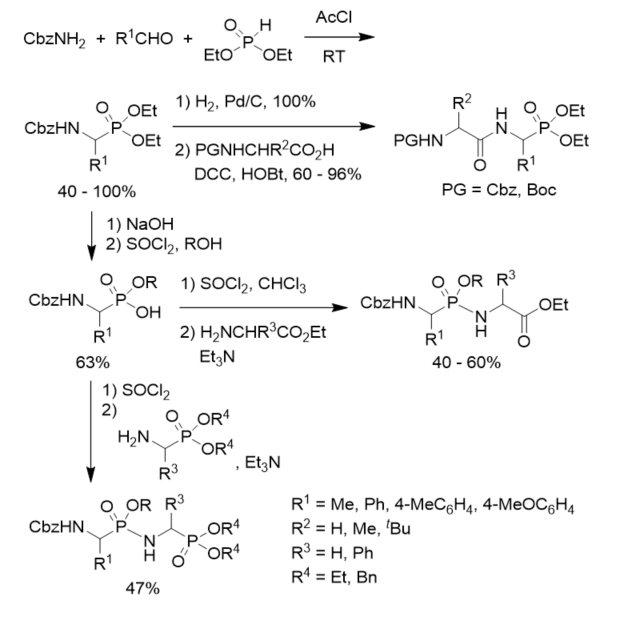
Scheme 6. Synthesis of phosphonopeptides composing of one or two 1-aminoalkylphosphonic acid residue(s).
To prepare novel inhibitors of VanX, a Zn(II) metalloenzyme that was required for high-level vancomycin resistance in bacteria, N-[(1-aminoethyl)hydroxyphosphinyl]-d-alanine was synthesized via coupling of N-Cbz protected methyl 1-aminoethylphosphonochloridate with methyl d-alaninate followed by basic hydrolysis and hydrogenolysis (Scheme 7). Bioassay results indicated that phosphonodipeptide was shown to be a partial competitive inhibitor of VanX with a Ki of 36 μM [32].
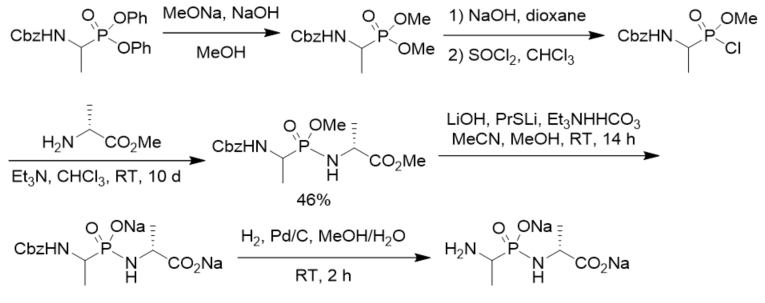
Scheme 7. Synthesis of phosphonopeptide inhibitors of VanX.
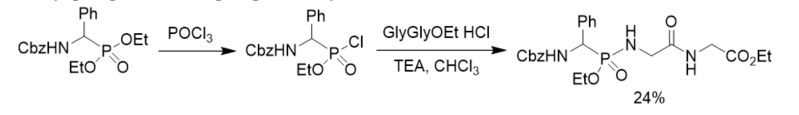
Phosphonopeptides CbzNHCH2P(O)(OEt)-l-ProOBn, CbzNHCH2P(O)(OEt)-d-Pro, CbzNHCH2P(O)(OEt)-l-thioProOMe, and H2NCH2P(O)(OEt)-l-Pro were prepared following similar method for investigation on the relative catalytic efficiency of β-lactamase catalyzed acyl and phosphyl transfer [33]. O-Phenyl phosphonamidates have been designed to bind covalently by nucleophilic substitution to the serine residue in the active site of serine proteases. Phosphonodipeptide with O-phenyl on the phosphorus atom was synthesized from phenyl N-Cbz 1-amino(phenyl)methylphosphonochloridate and methyl l-valinate. The stability of the phosphonamidates as a model of phosphonopeptides in aqueous solutions and their selectivity in the reaction against alcohols vs. thiols proved that they constituted a class of potential inhibitors of serine proteases and valuable tools to study the mechanism of inhibition. The transesterification of the phosphonopeptide phenyl ester with MeOH in the presence of Et3N/KF gave the corresponding phosphonopeptide methyl ester (Scheme 8) [34].
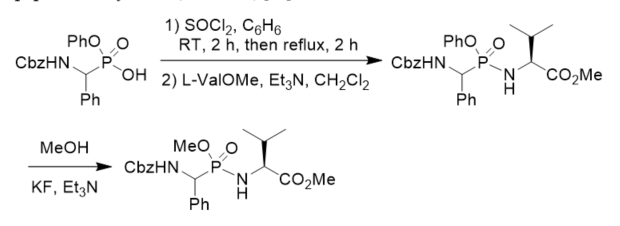
Scheme 8. Synthesis of phosphonopeptide and its transesterification.
By using similar synthetic method, this research group also prepared phosphonodipeptides as new inhibitors of leucine aminopeptidase [35] and investigated the influence of the nature of the N-terminal functional group on their hydrolysis [36].
Besides Cbz protecting group, Fmoc group was also used in the synthesis of phosphonopeptides. Benzyl hydrogen α-(9-fluorenylmethoxycarbonyl(Fmoc)amino)alkylphosphonates were obtained by the sodium metaperiodate oxidation of the corresponding phosphinates and converted to the phosphonochloridates, which were coupled with Nε-protected lysine benzyl ester to afford phosphonodipeptides containing a lysine residue (Scheme 9) [37].
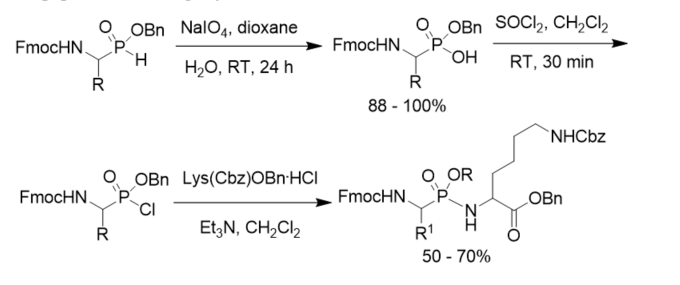
Scheme 9. Synthesis of N-Fmoc protected phosphonopeptides.
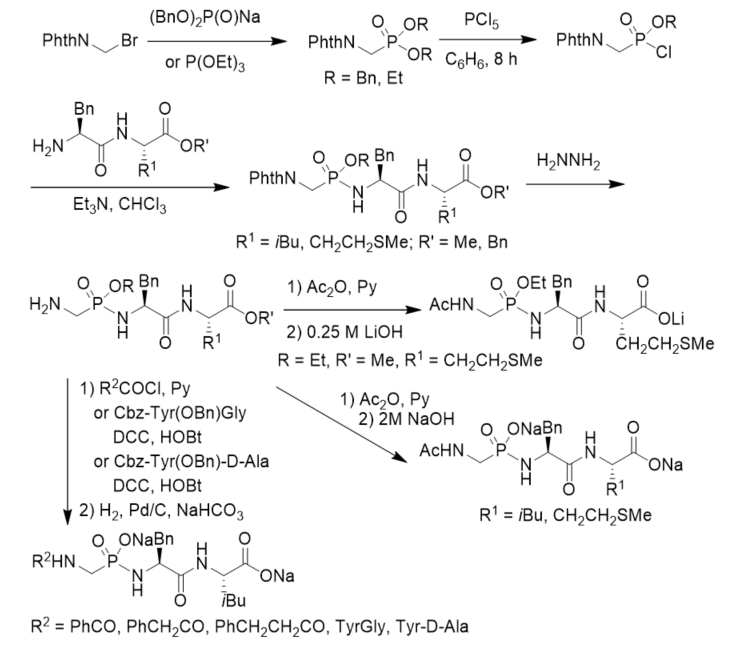
Cramer and Klebe described a procedure for the synthesis and purification of functionalized phosphonopeptides that were able to generate inhibitors for the metalloprotease thermolysin for use in biophysiological experiments. The method utilized an allyl ester/allyloxycarbonyl(Aloc) protection strategy and showed advantage of a fast and effective solid-phase purification step. They first prepared diallyl N-Cbz aminomethylphosphonate and transformed it into allyl N-Cbz aminomethylphosphonochloridate, which was further reacted with dipeptide allyl esters or amino amide derivatives. After deprotection under the catalysis of Pd(PPh3)4 followed by treatment with LiOH, phosphonopeptide lithium salts were obtained (Scheme 10). By using the strategy, they synthesized a series of highly polar phosphonopeptide inhibitors with amino- and hydroxy-functionalized side chains in excellent purity [38].
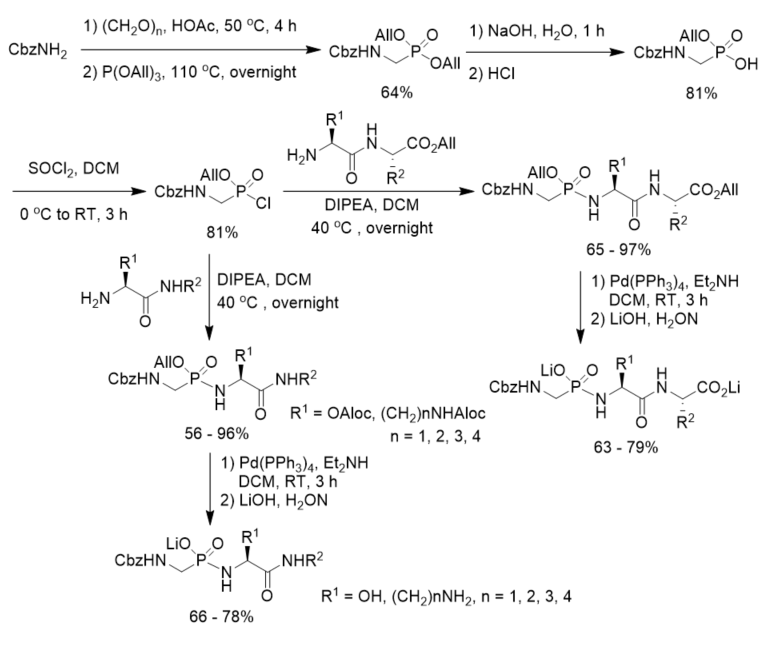
Scheme 10. Synthesis of phosphonopeptides from diallyl N-Cbz aminomethylphosphonate.
To search for effective and stable inhibitors of cytosolic leucine aminopeptidase, a β-phosphonopeptide containing aromatic N-terminal amino group was designed and synthesized. Diethyl 2-nitrophenylphosphonate was prepared and converted to the corresponding phosphonochloridate, which was coupled with methyl glycinate hydrochloride followed by reduction and basic hydrolysis, affording β-phosphonopeptide. The decrease in basicity of the terminal amino moiety of the β-phosphonopeptide resulted in satisfactory improvement of hydrolytic stability of the P-N bond. However, it did not exhibit inhibition activity up to millimolar concentration in enzymic assays towards leucine aminopeptidase possibly because diminishing the basic character of the terminal amino group resulted in a change of its affinity towards the zinc ions in the aminopeptidase. On the other hand, the decrease of the inhibition activity might also be ascribed to the steric constrain induced by the planar phenyl ring (Scheme 11) [39].
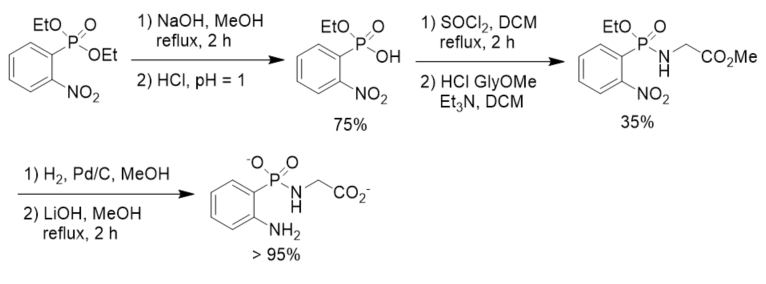
Scheme 11. Synthesis of β-phosphonopeptide containing aromatic N-terminal amino group.
2.4. Chlorination of Alkyl Phosphonic Acid Monoesters with Oxalyl Chloride
Moroder’s research group tested different chlorination conditions and found that oxalyl chloride-mediated preparation of phosphonochloridates allowed the improvement of the synthesis of phosphonopeptides, in comparison with thionyl chloride. A catalytic amount of N,N-dimethylformamide (DMF) promoted the chloridation. Particularly if AgCN was used as catalyst or upon conversion of the corresponding phosphonochloridates into their 7-aza-1-hydroxybenzotriazole (HOAt) ester, the yields of phosphonopeptides even reached 90% (Scheme 12). When peptide hydrochlorides were utilized as amino components triethylamine or diisoproylethylamine was added as base. However, triphosgene was an inefficient chloridating reagent. Furthermore, the Mukaiyama procedure failed completely in the present case. The mixed anhydrides of aminoalkylphosphonic acid monoesters with pivaloyl chloride generated, but affording exclusively to the N-pivaloylpeptide derivatives instead phosphonopeptides, despite the steric bulk of the tert-butyl group.
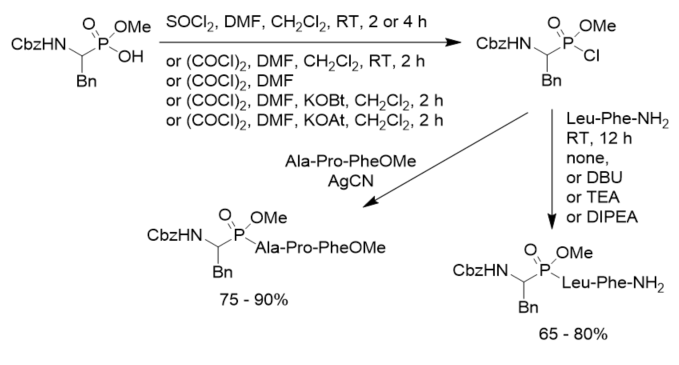
Scheme 12. Synthesis of phosphonopeptides under mild conditions.
Various methods have been explored for the convergent synthesis of phosphonopeptides via phosphonochloridates. PCl5 is an effective reagent for the conversion of diethyl and diisopropyl phosphonates to the corresponding phosphonochloridates in the absence of complex functionality. Subsequent reaction with amino and peptide esters generated the phosphonopeptides in good yields. However, a milder method using oxalyl chloride to generate the phosphonochloridates from phosphonic monoesters was required in the presence of more complex functionality. Aminolysis of the dimethyl phosphonates or basic hydrolysis of diethyl phosphonates generated the requisite phosphonic monoesters for the preparation of phosphonochloridates [40].
New muramyl dipeptide (MDP) analogs related to LK 423 as potential immunomodulators was synthesized by coupling of methyl N-Fmoc 1-aminoethylphosphonochloridate and dibenzyl d-glutamate toluenesulfonate, followed by basic deprotection and coupling with 5-phthalimidopentanoic acid. The dipeptide part of the lead compound was modified by introducing a phosphonamidate bond instead of the amide bond between l-alanine and d-glutamic acid (Scheme 13) [25].
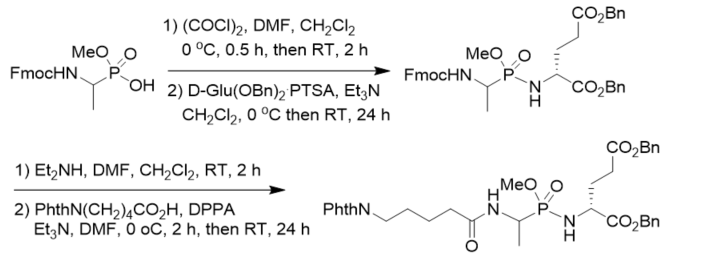
Scheme 13. Synthesis of phosphonopeptides as potential immunomodulators.
The synthesis of different muramyl dipeptide analogue LK 415 derivatives as potential immunomodulators was reported. The d-alanine and d-isoglutamine of LK 415 were replaced by their phosphonic analogues l,d-phosphonoalanine [H2NCHMeP(O)(OMe)2] and H2NCH[CH2CH2P(O)(OEt)2]COCH2Ph, yielding the LK 415 analogues N-[2-[2-[(1-adamantylcarbonyl)amino]ethoxy]acetyl]-NHCHMeP(O)(OMe)-d-Glu(OEt)2 and N-[2-[2-[(1-adamantylcarbonyl)amino]ethoxy]acetyl]-l-Ala-NHCH[CH2CH2P(O)(OEt)2]COCH2Ph, respectively [41].
To prepare phosphorus analogues of γ-glutamyl peptide, N-Cbz l-glutamic acid was first transformed to the corresponding dimethyl phosphonate. After aminolysis and chlorination it was transformed to the corresponding phosphonochloridate, which was reacted with diethyl glutamate, affording γ-phosphonodipeptide in 64% yield. After hydrogenolysis, N-terminal free γ-phosphonopeptide was obtained in 87% yield. However, the coupling of the phosphonochloridate with diethyl 2-hydroxyglutarate generated γ-phosphonodepsipeptide in only 6.7% yield, indicating the strategy was not suitable for the synthesis of γ-phosphonodepsipeptide (Scheme 14)[42].
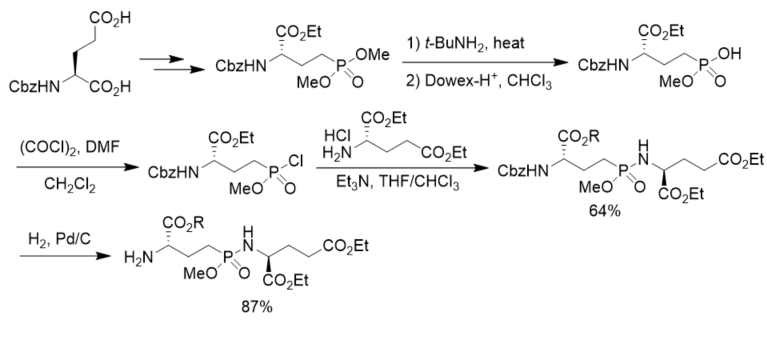
Scheme 14. Synthesis of γ-phosphonopeptides.
2.5. Chlorination of Alkyl Trimethylsilyl Phosphonites or Alkyl Phosphinates with Carbon Tetrachloride
To minimize the side reactions (formation of oxazaphospholines) of N-Cbz protected 1-aminoalkylphosphonochloridates, a convenient method for the synthesis of phosphonopeptides was described. N-Cbz protected 2-aminoalkylphosphinates were converted to their trimethylsilyl phosphonite tautomers with the treatment with bis(trimethylsilyl)acetamide. They were oxidized with CCl4 to generate the corresponding phosphonochloridates as intermediates, which reacted with amino acid esters to give rise to the desired phosphonopeptides. The oxidative activation was carried out in the presence of the amine nucleophiles so that stoichiometric formation of the phosphonochloridates were avoided and side reactions were minimized (Scheme 15) [26].
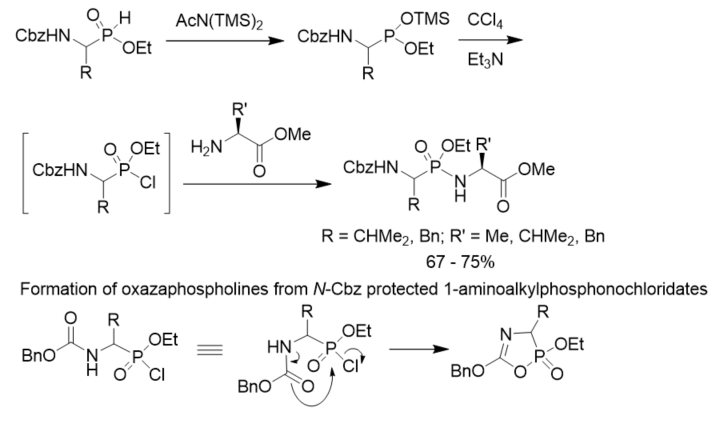
Scheme 15. Synthesis of phosphonopeptides via chlorination of aminoalkylphosphonites.
Phosphonylation of amino acid esters with N-Cbz 1-aminoalkylphosphonochloridates was a general method for the preparation of phosphonopeptides containing a phosphonamidate bond. N-Cbz 1-aminoalkylphosphonochloridates were prepared from both N-Cbz 1-aminoalkylphosphinates with carbon tetrachloride in the presence of trimethylamine through the Atherton–Todd reaction and the corresponding N-Cbz 1-aminoalkylphosphonic acid monoesters with thionyl chloride (Scheme 16) [27].
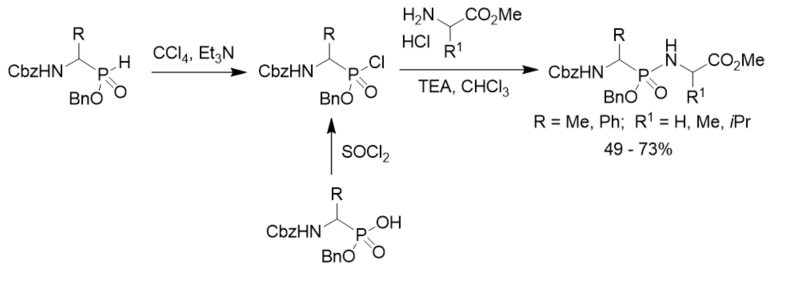
Scheme 16. Synthesis of phosphonopeptides from aminoalkylphosphinates.
2.6. Bromination of Alkyl Phosphinates with Bromine
Yao and Yuan developed a general and efficient one-pot procedure for converting 1,1-diethoxyalkylphosphinates into phosphonates or phosphonamides by the use of bromine. For α-aminoalkylphosphinates, the transformation could be realized without the protection of the amino group. Enantiopure phosphinate reacted stereospecifically with bromine and subsequently couples with nucleophile to generate the corresponding optically active derivatives with retention of configuration at the phosphorus center (Scheme 17) [28]. The developed method was applied in the synthesis of phosphonopeptides.
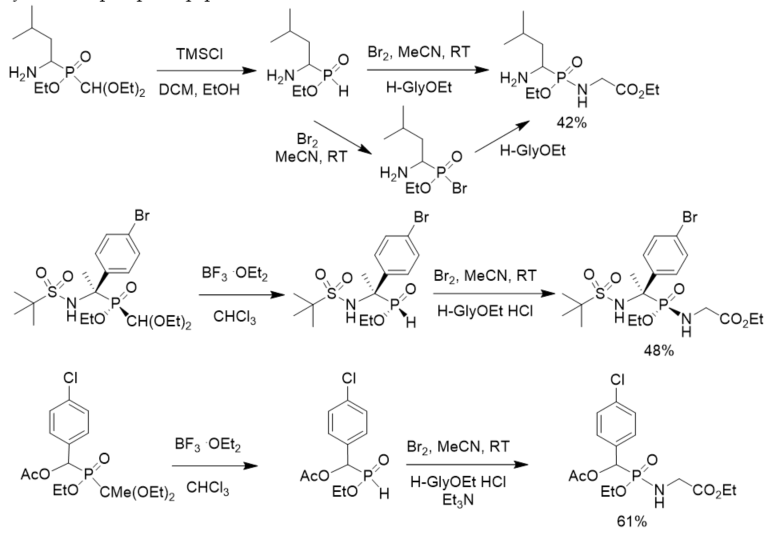
Scheme 17. Synthesis of phosphonopeptides via phosphonobromidates.
A new method for the synthesis of tert-butoxycarbonyl(Boc)-protected phosphonopeptides was developed with diethyl 1-azidoalkylphosphonates as starting materials. After partial hydrolysis and treatment with oxalyl chloride under the catalysis of DMF, diethyl 1-azidoalkylphosphonates were transformed to ethyl 1-azidoalkylphosphonochloridates, which were coupled with amino ester hydrochlorides to afford azidophosphonodipeptides. They were further converted N-Boc protected phosphonodipeptides under hydrogenolysis in the presence of Boc2O in ethyl acetate (Scheme 18) [43].
The click reaction of azidophosphonodipeptides and N-protected leucine/isoleucine propargyl esters with different protecting group (PG) under the catalysis of copper sulfate pentahydrate, affording triazole-containing phosphonopeptides (Scheme 18) [44].
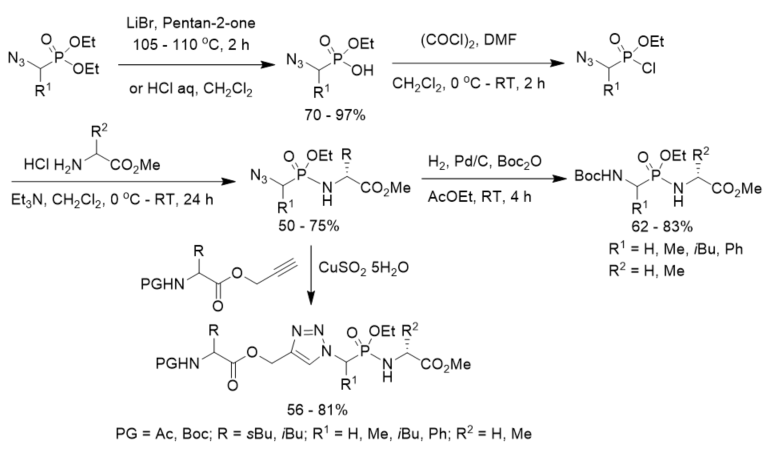
Scheme 18. Synthesis of phosphonopeptides and triazole-containing phosphonopeptides via 1-azidoalkylphosphonochloridates.
References
- Kafarski, P.; Lejczak, B. Synthesis of Phosphono- and Phosphinopeptides in Aminophosphonic and Aminophosphinic Acids; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: West Sussex, England, 2000; pp. 173–203.
- Kafarski, P.; Lejczak, B. The Biological Activity of Phosphono- and Phosphinopeptides in Aminophosphonic and Aminophosphinic Acids; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: West Sussex, England, 2000; pp. 407–442.
- Allen, J.G.; Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Nisbet, L.J.; Ringrose, P.S. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58.
- Bartlett, P.A.; Marlowe, C.K. Evaluation of intrinsic binding energy from a hydrogen bonding group in an enzyme inhibitor. Science 1987, 235, 569–571.
- Krimmer, S.G.; Cramer, J.; Betz, M.; Fridh, V.; Karlsson, R.; Heine, A.; Klebe, G. Rational design of thermodynamic and kinetic binding profiles by optimizing surface water networks coating protein-bound ligands. J. Med. Chem. 2016, 59, 10530–10548.
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity J. Med. Chem. 2003, 46, 2641–2655.
- Yang, K.W.; Cheng, X.; Zhao, C.; Liu, C.C.; Jia, C.; Feng, L.; Xiao, J.M.; Zhou, L.S.; Gao, H.Z.; Yang, X.; et al. Synthesis and activity study of phosphonamidate dipeptides as potential inhibitors of VanX. Bioorg. Med. Chem. Lett. 2011, 21, 7224–7227.
- Demange, L.; Moutiez, M.; Dugave, C. Synthesis and evaluation of Glyψ(PO2R-N)Pro-containing pseudopeptides as novel inhibitors of the human cyclophilin hCyp-18. J. Med. Chem. 2002, 45, 3928–3933.
- Schultz, P.G.; Lerner, R.A. Antibody catalysis of difficult chemical transformations. Acc. Chem. Res. 1993, 26, 392–395.
- Jacobsen, J.R.; Schultz, P.G. Antibody catalysis of peptide-bond formation. Proc. Nat. Acad. Sci. USA 1994, 91, 5888–5892.
- Xie, G.Y.; Chen, J.H.; Lin, S.S.; Jin, S. The synthesis of a novel phosphorus containing antigen. Chin. Chem. Lett. 2001, 12, 497–498.
- Xie, G.Y.; Chen, L.M.; Chen, J.H. Synthesis of a novel antigen containing phosphorus. Chem. J. Chin. Univ. 2003, 24, 1037–1039.
- Hariharan, M.; Chaberek, S.; Martell, A.E. Synthesis of phosphonopeptide derivatives. Synth. Commun. 1973, 3, 375–379.
- Jackson, D.S.; Fraser, S.A.; Ni, L.-M.; Kam, C.-M.; Winkler, U.; Johnson, D.A.; Froelich, C.J.; Hudig, D.; Powers, J.C. Synthesis and evaluation of diphenyl phosphonate esters as inhibitors of the trypsin-like granzymes A and K and mast cell tryptase. J. Med. Chem. 1998, 41, 2289–2301.
- Cervera-Villanueva, J.M. Jr.; Viveros-Ceballos, J.L.; Linzaga-Elizalde, I.; Ordonez, M. Practical synthesis of novel phosphonopeptides containing AibP. J. Peptide Sci. 2016, 22, 70–75.
- Kudzin, Z.H.; Depczynski, R.; Kudzin, M.H.; Drabowicz, J. 1-(N-chloroacetylamino)-alkylphosphonic acids—synthetic precursors of phosphonopeptides. Amino Acids 2008, 34, 163–168.
- Sun, B.Y.; Xu, J.X. Convenient synthesis of phosphonodipeptides containing C-terminal -aminoalkylphosphonic acids. J. Pept. Sci. 2015, 21, 615–619.
- Sun, B.Y.; Xu, J.X. Convenient and facile synthesis of hybrid sulfonophosphonodipeptides. Synthesis 2015, 47, 1479–1487.
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910.
- Elliott, R.L.; Marks, N.; Berg, M.J.; Portoghese, P.S. Synthesis and biological evaluation of phosphonamidate peptide inhibitors of enkephalinase and angiotensin-converting enzyme. J. Med. Chem. 1985, 28, 1208–1216.
- Xu, J.X.; Xia, C.F.; Yu, L.; Zhou, Q.Z. Synthesis of phosphonopeptides containing 1-aminoalkylphosphonic acid. Phosphorus Sulfur Silicon Relat. Elem. 1999, 152, 35–44.
- Vo Quang, Y.; Gravey, A.M.; Simonneau, R.; Vo Quang, L.; Lacoste, A.M.; Le Goffic, F. Towards new inhibitors of D-alanine:D-alanine ligase: The synthesis of 3-aminobutenylphosphonic and aminophosphonamidic acids. Tetrahedron Lett. 1987, 28, 6167–6170.
- Lacoste, A.M.; Chollet-Gravey, A.M.; Vo Quang, L.; Vo Quang, Y.; Le Goffic, F. Time-dependent inhibition of Streptococcus faecalis D-alanine:D-alanine ligase by α-aminophosphonamidic acids. Eur. J. Med. Chem. 1991, 26, 255–260.
- Musiol, H.-J.; Grams, F.; Rudolph-Boehner, S.; Moroder, L. The chemistry of phosphapeptides: Formation of functionalized phosphonochloridates under mild conditions and their reaction with alcohols and amines. On the synthesis of phosphonamidate peptides. J. Org. Chem. 1994, 59, 6144–6146.
- Gobec, S.; Urleb, U. Synthesis of new lipophilic phosphonate and phosphonamidate analogs of N-acetylmuramyl-D-isoglutamine related to LK 423. Molecules 2002, 7, 394–404.
- Sampson, N.S.; Bartlett, P.A. Synthesis of phosphonic acid derivatives by oxidative activation of phosphinate esters. J. Org. Chem. 1988, 53, 4500–4503.
- Mucha, A.; Kafarski, P.; Plenat, F.; Cristau, H.-J. The preparation of phosphono peptides containing a phosphonamidate bond. Tetrahedron 1994, 50, 12743–12754.
- Yao, Q.L.; Yuan, C.Y. Stereospecific transformation of protected PH group into PO or PN group in one-pot reaction. J. Org. Chem. 2012, 77, 10985–10990.
- Horiguchi, M.; Kandatsu, M. Isolation of 2-aminoethane phosphonic acid from Rumen Protozoa. Nature 1959, 184, 901–902.
- Kittredge, J.S.; Roberts, E.; Simonsen, D.G. The occurrence of free 2-aminoethylphosphonic acid in the sea anemone, Anthopleura elegantissima. Biochemistry 1962, 1, 624–628.
- Yuan, C.Y.; Wang, G. H Studies on organophosphorus compounds. 65. A facile synthetic route to phosphonopeptides. Phosphorus Sulfur Silicon Relat. Elem. 1992, 71, 207–212.
- Yang, K.-W.; Brandt, J.J.; Chatwood, L.L.; Crowder, M.W. Phosphonamidate and phosphothioate dipeptides as potential inhibitors of VanX. Bioorg. Med. Chem. Lett. 2000, 10, 1085–1087.
- Slater, M.J.; Laws, A.P.; Page, M.I. The relative catalytic efficiency of β-lactamase catalyzed acyl and phosphyl transfer. Bioorg. Chem. 2001, 29, 77–95.
- Mucha, A.; Kafarski, P. Transesterification of monophenyl phosphonamidates-chemical modeling of serine protease inhibition. Tetrahedron 2002, 58, 5855–5863.
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. Structure-based design and synthesis of dipeptide analogs as new inhibitors of leucine aminopeptidase. Phosphorus Sulfur Silicon Relat. Elem. 2002, 177, 1739–1743.
- Mucha, A.; Grembecka, J.; Cierpicki, T.; Kafarski, P. Hydrolysis of the phosphonamidate bond in phosphono dipeptide analogs—the influence of the nature of the N-terminal functional group. Eur. J. Org. Chem. 2003, 4797–4803, doi:10.1002/ejoc.200300469.
- Dumy, P.; Escale, R.; Girard, J.P.; Parello, J.; Vidal, J.P. A convenient synthetic approach to new α-(9-fluorenylmethoxycarbonylamino)alkylphosphonic acid derivatives. Synthesis 1992, 1226–1228, doi: 10.1055/s-1992-26344.
- Cramer, J.; Klebe, G. An allyl protection and improved purification strategy enables the synthesis of functionalized phosphonamidate peptides. Synthesis 2017, 49, 1857–1866.
- Mucha, A.; Kunert, A.; Grembecka, J.; Pawelczak, M.; Kafarski, P. A phosphonamidate containing aromatic N-terminal amino group as inhibitor of leucine aminopeptidase-design, synthesis and stability. Eur. J. Med. Chem. 2006, 41, 768–772.
- Malachowski, W.P.; Coward, J.K. The chemistry of phosphapeptides: Formation of functionalized phosphonochloridates under mild conditions and their reaction with alcohols and amines. J. Org. Chem. 1994, 59, 7616–7624.
- Urleb, U.; Gobec, S. Synthesis of new adamantane substituted acyclic MDP analogs related to LK 415. Acta Pharm. 2000, 50, 173–184.
- Malachowski, W.P.; Coward, J.K. The chemistry of phosphapeptides: Investigations on the synthesis of phosphonamidate, phosphonate, and phosphinate analogs of glutamyl-γ-glutamate. J. Org. Chem. 1994, 59, 7625–7634.
- Sikora, D.; Nonas, T.; Gajda, T. O-Ethyl 1-azidoalkylphosphonic acids-versatile reagents for the synthesis of protected phosphonamidate peptides. Tetrahedron 2001, 57, 1619–1625.
- Artyushin, O.I.; Sharova, E.V.; Yarkevich, A.N.; Genkina, G.K.; Vinogradova, N.V.; Brel, V.K. Design of phosphonate analogs of short peptides by “Click” chemistry. Russ. Chem. Bull. 2015, 64, 2172–2177.


