 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Worapon Kiatkittipong | + 334 word(s) | 334 | 2020-08-21 03:16:07 | | | |
| 2 | Worapon Kiatkittipong | -3 word(s) | 331 | 2020-08-24 18:51:50 | | | | |
| 3 | Worapon Kiatkittipong | -3 word(s) | 331 | 2020-08-24 18:54:12 | | | | |
| 4 | Bruce Ren | + 4852 word(s) | 5183 | 2020-08-27 04:05:46 | | | | |
| 5 | Bruce Ren | + 4852 word(s) | 5183 | 2020-08-27 04:25:55 | | |
Video Upload Options
In the present review (10.3390/pr8050548), CO2 cycloaddition can be seen as a reasonably competent alternative to CO2 transformation, offsetting the high value-added nature by extending material use defer CO2 back to the atmosphere when compared to commodities and fuels such as urea, methanol, and methane.
Abstract
Carbon dioxide (CO2) has been anticipated as an ideal carbon building block for organic synthesis due to the noble properties of CO2, which are abundant renewable carbon feedstock, non-toxic nature, and contributing to a more sustainable use of resources. Several green and proficient routes have been established for chemical CO2 fixation. Among the prominent routes, this review epitomizes the reactions involving cycloaddition of epoxides with CO2 in producing cyclic carbonate. Cyclic carbonate has been widely used as a polar aprotic solvent, as an electrolyte in Li-ion batteries, and as precursors for various forms of chemical synthesis such as polycarbonates and polyurethanes. This review provides an overview in terms of the reaction mechanistic pathway and recent advances in the development of several classes of catalysts, including homogeneous organocatalysts (e.g., organic salt, ionic liquid, deep eutectic solvents), organometallic (e.g., mono-, bi-, and tri-metal salen complexes and non-salen complexes) and heterogeneous supported catalysts, and metal organic framework (MOF). Selection of effective catalysts for various epoxide substrates is very important in determining the cycloaddition operating condition. Under their catalytic systems, all classes of these catalysts, with regard to recent developments, can exhibit CO2 cycloaddition of terminal epoxide substrates at ambient temperatures and low CO2 pressure. Although highly desired conversion can be achieved for internal epoxide substrates, higher temperature and pressure are normally required. This includes fatty acid-derived terminal epoxides for oleochemical carbonate production. The production of fully renewable resources by employment of bio-based epoxy with biorefinery concept and potential enhancement of cycloaddition reactions are pointed out as well.
1. Introduction
The significant concern with regard to the environmental impact of anthropogenic CO2 emission into the atmosphere has directly contributed to global warming, thus demanding the need for mitigating CO2 emission [1]. Avoidance of CO2 emission through improved energy and material efficiency and the use of renewable energy and material would be the most desirable carbon management strategy. After this, utilization of CO2 by extending material use, referred to as “carbon capture and utilization (CCU)”, for making repeated use of emitted CO2, would be beneficial, followed by carbon capture and storage (CCS) for long-term geological sequestration of inevitable residual CO2 [2]. CCU is emphasized as an adjunct, not alternative, to CCS [3]. There are many diverse approaches for CO2 utilization; however, they generally can be divided into two main approaches: (1) direct use, and (2) transformation via chemical and biological processes, as depicted in Figure 1.
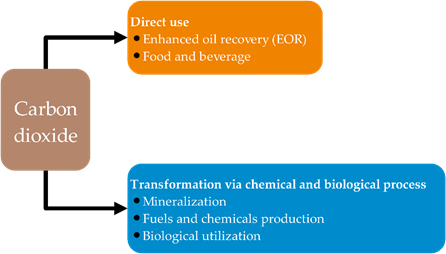
Figure 1. The approaches to utilize carbon dioxide.
CO2 direct uses have been found in oil and gas industries, as well as in food and beverages such as carbonated drinks. Presently, the major CO2 utilization is based on a direct use of CO2 in the oil and gas industries, which mainly relies on enhanced oil recovery (EOR) or other related technologies, such as enhanced coal-bed methane recovery (ECBM) and enhanced shale gas recovery (ESGR). It is worth nothing that, considering the carbon life cycle, these enhanced production of fossil fuel technologies still produce a surplus of CO2 emissions. Readers are referred to a current review performed by Zhang et al. for more insightful information. For the second approach, transformation via chemical and biological process includes mineralization, fuel and chemical production, and biological utilization. Although these CO2 transformations cannot mitigate the enormous CO2 emissions, converting CO2 to chemicals has emerged and has drawn much research attention because it offers extending material use with higher value. CO2 has been foreseen as a carbon building block for organic syntheses, as CO2 is renewable, non-toxic, and economical [4]. However, the thermodynamically stable and kinetic inertness of CO2 has hindered CO2 activation and fixation [5]. The enhancement of efficient chemical processes for the chemical fixation of CO2 into high value-added organic chemicals should be vigorously developed. Several proficient routes have been established for chemical CO2 fixation [6]. However, in general, only several processes have been commercialized because of limitation in terms of precursor and efficiency of the reactions due to the requirement of reactive agents for CO2 activation (Figure 2) [7][8][9].
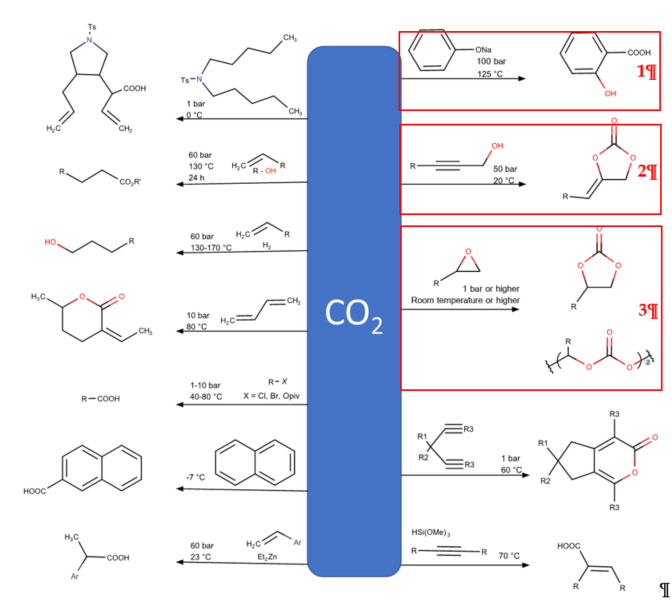
Figure 2. Examples of CO2 as C1 feedstock in organic synthesis. The synthesis reactions marked with red boxes have been industrialized[7][8][9].
The Kolbe–Schmitt reaction (Figure 2; red box number 1) is one of the most essential and renowned carboxylation reactions, offering an economical pathway to produce salicylic acids by carboxylation of phenoxides with CO2 [10]. Salicylic acids are crucial chemicals in pharmaceuticals and agrochemicals, including vital precursors in organic synthesis [11]. Because of the low cost of the reaction, the Kolbe–Schmitt reaction is most stable and extensively used in industry, especially in the synthesis process of aspirin. Consequently, there is no considerable change that can be made to improve this reaction. In addition, the reaction of propargylic alcohol with CO2 to produce cyclic carbonate with the presence of base is applicable in industry (Figure 2; red box number 2) [12]. However, its industrial applicability on a larger scale is limited because of salt waste formation due to the need of the acid addition to form free carboxylic acid [13].
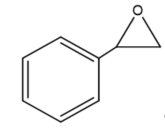
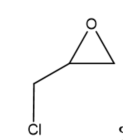
This review focuses on the most prominent route, which is the cycloaddition of epoxides into CO2, producing cyclic carbonate and polymeric carbonate (Figure 2; red box number 3) [14]. The five-membered ring cyclic carbonates are extensively utilized as sustainable polar aprotic solvents, value-added compound for fuel, and electrolytes for lithium-ion batteries, and are the precursor for polymerization reactions and pharmaceuticals [15]. The CO2 coupling with epoxide captivates the interest of industries because of readily available precursors and widely used products that are cyclic carbonates . Figure 3 shows typical and some emerging epoxide species, which can be classified as terminal epoxides (Figure 3a–g) and internal epoxides (Figure 3h–i). Their structure and abbreviations used in this manuscript are provided.
Ethylene carbonate and propylene carbonate are the most significant cyclic carbonates employed in industry. They are commercially catalyzed by quaternary ammonium or phosphonium salts, which are relatively inefficient, and thus require high temperature and pressure [16]. For the past decades, numerous catalysts, either homogeneous or heterogeneous, therefore have been extensively investigated.
This paper aims to review recent advances and trends in catalytic CO2 cycloaddition, including homogeneous organocatalyst (e.g., organic salt, ionic liquid, deep eutectic solvents), organometallic (e.g., mono-, bi-, and tri-metal salen complexes and non-salen complexes), and heterogeneous supported catalyst and metal organic frameworks (MOFs), along with the reaction mechanistic pathway. Substitution with bio-based epoxy is a proposed representation of pathways via the amorphous sugar, lipid, and lignocellulosic biomass platform. Last but not least, the possibility of enhancing cycloaddition reactions through emerging strategies is discussed.
|
Terminal epoxides: |
|
|
(a) |
(b) |
(c) |
(d) |
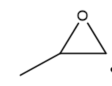
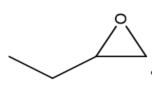
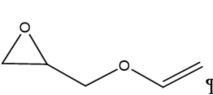
|
(e) |
(f) |
(g) |
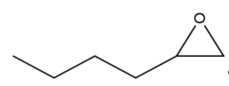
|
Internal epoxides: |
|
|
(h) |
(i) |
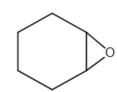
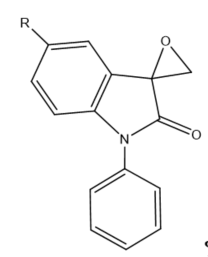
Figure 3. Structure of typical and some emerging epoxide species: (a) ethylene oxide (EO); (b) propylene oxide (PO); (c) butylene oxide (BO); (d) allyl glycidyl ether (AGE); (e) hexene oxide (HO); (f) styrene oxide (SO); (g) epichlorohydrin (ECH); (h) cyclohexene oxide (CHO); (i) spiro-epoxy oxindole (SEO).
2. Cycloaddition Reaction Mechanism of Epoxide with CO2
The reaction involving cycloaddition of CO2 with epoxides can be instigated via activating of either the CO2 or epoxide, or both concurrently [17]. Figure 4 illustrates the general mechanistic pathway of CO2 cycloaddition reaction. The CO2 activation can take place either through a nucleophilic (Nu) attack byoxygen atom to act as a nucleophile (cycle 1, first step) or an electrophilic attack by carbon atom to act as an electrophile (cycle 2, second step). The epoxide can be activated readily by interacting the oxygen atom with Lewis acid (LA), followed by a nucleophilic attack, promoting epoxide ring opening (cycle 2, first step). Thus, most of the catalytic systems used for CO2 addition into epoxides contain Lewis acid sites (LA) for the later electrophilic activation of epoxide [18].
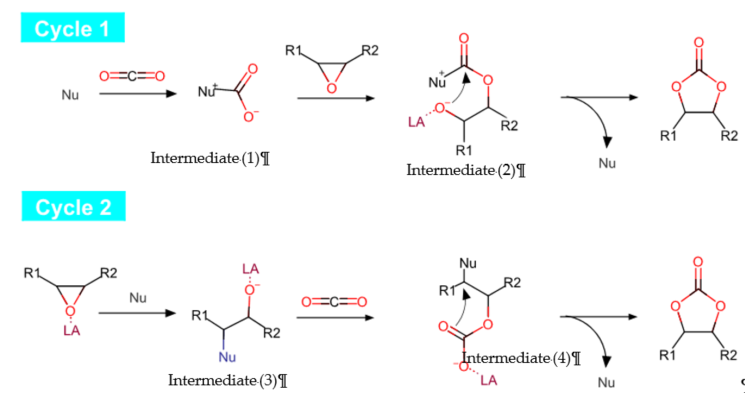
Figure 4. Reaction mechanistic pathways of cycloaddition of epoxide with CO2.
Cycle 1 demonstrates the activation of CO2 through nucleophilic attack to form an intermediate (1), which later promotes epoxide ring opening (the presence of Lewis acid (LA) site may facilitate the ring opening), leading to the second intermediate (2). Then, cyclization occurs to produce a cyclic carbonate while the nucleophile is recycled for a new catalytic cycle. Furthermore, cycle 2 demonstrates a commonly proposed pathway through activation of epoxide. The Lewis base compounds, such as quaternary ammonium halide salts and phosphonium salts [16], acting as nucleophiles attack the epoxide to enable ring opening, producing an intermediate (3), which may be assisted by a Lewis acid (LA) species in proximity to metal centers. Subsequently, upon ring opening, the CO2 insertion occurs in the metal–alkoxide bond of the intermediate, producing a new intermediate (4). The cyclization of the intermediate produces cyclic carbonate as the final product, and nucleophile is recycled for further reaction.
3. Catalysts for Cycloaddition of Epoxide with CO2
Figure 5 displays the classification of catalyst for cycloaddition of epoxide with CO2. The contents are outlined following the classification.
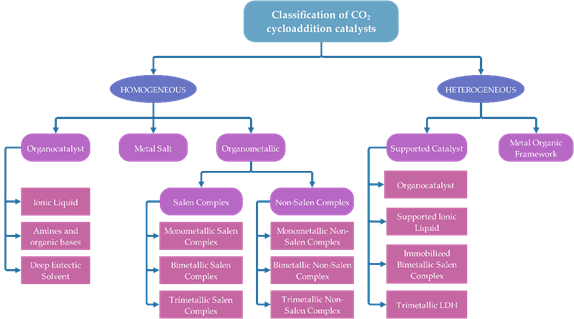
Figure 5. Classification of catalysts for cycloaddition of epoxide with CO2.
3.1. Homogeneous Catalyst
Homogeneous catalysis generally involves the catalyst and reactant presenting together in the same phase (generally liquid). Homogeneous catalytic systems usually display the most active high conversion and selectivity. On the contrary, for the heterogeneous catalytic system, the catalyst is usually in a solid form while the reactant can be in liquid or gas phases. Usually, the heterogeneous catalyst is less active when compared with their homogeneous analogues because the limitation of the reactant to migrate to the catalytic sites causes low activity of heterogeneous catalysts, originating from poor diffusion . However, homogeneous catalysts have several practical disadvantages, namely, lesser robust and lower recyclability as opposed to heterogeneous catalysts. One of the most use techniques for combining the high activity of a homogeneous catalyst with easily and effectively catalyst recovery is the “heterogenization” of the active species via such functionalization/immobilization support materials [19].
Table 1 summarizes different classes and types of homogeneous catalysts on CO2 cycloaddition reaction performance.
Table 1. Operational conditions and the corresponding outputs from the CO2 cycloaddition reactions catalyzed by different homogeneous catalysts.[20][21][22][23][24][25][26]
|
Epoxide |
Catalyst Class/Type |
Cocatalyst |
T (℃) |
P (bar) |
Time (h) |
Yield (%) |
Conv. (%) |
Ref. |
|
|
|
Organocatalysts |
|
|||||||||
|
ECH |
Ionic liquid/2,6-pyridinedimethanol |
TBACl |
25 |
1.01 |
24 |
52 |
ND |
[14] |
|
|
|
TBAB |
25 |
1.01 |
24 |
67 |
ND |
|
||||
|
TBAI |
25 |
1.01 |
24 |
92 |
ND |
|
||||
|
SEO |
DES/ChCl:glycerol |
- |
40 |
1.01 |
5 |
20 |
ND |
[20] |
|
|
|
DES/ChCl:Ethylene glycol |
- |
40 |
1.01 |
5 |
10 |
ND |
|
|||
|
DES/ChCl:benzoic acid |
- |
40 |
1.01 |
5 |
12 |
ND |
|
|||
|
DES/ChCl:Urea(100mg) |
- |
40 |
1.01 |
5 |
49 |
ND |
|
|||
|
- |
60 |
1.01 |
5 |
85 |
ND |
|
||||
|
- |
70 |
1.01 |
5 |
89 |
ND |
|
||||
|
DES/ChCl:Urea (200mg) |
- |
70 |
1.01 |
3 |
93 |
ND |
|
|||
|
DES/ChCl:Urea (300mg) |
- |
70 |
1.01 |
2 |
98 |
ND |
|
|||
|
Monometallic salen complexes |
|
|||||||||
|
SO |
Salophen (Figure 6a; R = tert-butyl, X = Cl) |
TBAB |
25 |
1 |
3 |
≈37 a |
37 |
[21] |
|
|
|
|
|
|
6 |
≈60 a |
60 |
|
|
|||
|
24 |
≈100 a |
100 |
|
|||||||
|
Salophen (Figure 6a; R = MeO, X = Br) |
TBAB |
25 |
1 |
3 |
≈44 a |
44 |
|
|||
|
6 |
≈71 a |
71 |
|
|||||||
|
24 |
≈100 a |
100 |
|
|||||||
|
Bimetallic salen complexes |
|
|||||||||
|
PO |
Bimetallic Salen-Co |
TBAB |
25 |
10 |
48 |
75.8 |
ND |
|
||
|
PO |
Bimetallic Salen-Al |
TBAB |
25 |
10 |
48 |
73.2 |
ND |
|
|
|
|
PO |
Bimetallic Salen-Zn |
TBAB |
25 |
10 |
48 |
72.1 |
ND |
|
||
|
Monometallic non-salen complexes |
|
|||||||||
|
HO |
Zn(II) TPP |
- |
20 |
1 |
43 |
82 |
ND |
[23] |
||
|
PO |
Co-cryptand |
TBAB |
0 |
1.01 |
8 |
43 |
ND |
[24] |
|
|
|
SO |
TBAB |
20 |
1.01 |
12 |
32 |
ND |
|
|||
|
ECH |
TBAB |
20 |
1.01 |
24 |
48 |
ND |
|
|||
|
- |
20 |
1.01 |
48 |
6 |
ND |
|
||||
|
- |
TBAB |
20 |
1.01 |
48 |
11 |
ND |
|
|||
|
Bimetallic non-salen complexes |
|
|||||||||
|
SO |
Bi-aluminium scorpionate |
R=(S)-CH(PhMe; X=Et |
TBAB |
RT |
1 |
24 |
75.0 b |
77 |
[25] |
|
|
TBAB |
RT |
10 |
24 |
97.3 b |
100 |
|
||||
|
R=Ph; X=Me |
TBAB |
RT |
10 |
24 |
58.6 b |
60 |
|
|||
|
Trimetallic non-salen complexes |
|
|||||||||
|
SO |
Trinuclear aluminium scorpionate |
R=Bu; X=Et |
TBAB |
RT |
10 |
24 |
97.3 b |
100 |
[25] |
|
|
TBAB |
RT |
1 |
24 |
75.0 b |
77 |
|
||||
|
R=(S)-CH(Ph)Me; X=Me |
TBAB |
RT |
10 |
24 |
90.2 b |
92 |
|
|||
|
TBAB |
RT |
1 |
24 |
50.4 b |
52 |
|
||||
|
R=(S)-CH(Ph)Me; X=Et |
TBAB |
RT |
10 |
24 |
97.3 b |
100 |
|
|||
|
TBAB |
RT |
1 |
24 |
75.0 b |
77 |
|
||||
|
R=Ph; X=Me |
TBAB |
RT |
10 |
24 |
97.3 b |
100 |
|
|||
|
TBAB |
RT |
1 |
24 |
97.3 b |
100 |
|
||||
|
CHO |
Trimetallic-Co |
TBAB |
80 |
20.68 |
24 |
66.3 |
67 |
[26] |
|
|
|
TBAI |
80 |
20.68 |
24 |
38.6 |
39 |
|
||||
|
Trimetallic-Zn |
TBAB |
80 |
20.68 |
24 |
90.0 |
91 |
|
|||
|
TBAI |
80 |
20.68 |
24 |
87.1 |
88 |
|
||||
|
Trimetallic-Ni |
TBAB |
80 |
20.68 |
24 |
42.6 |
43 |
|
|||
Abbreviations: ChCl, choline chloride; Conv, conversion; ND, not determine; RT, room temperature; TBAB, tetrabutylammonium bromide; TBACl, tetrabutylammonium chloride; TBAI, tetrabutylammonium iodide. a Calculated from reported conversion with a selectivity higher than 99%, b Calculated from turnover frequency (TOF).
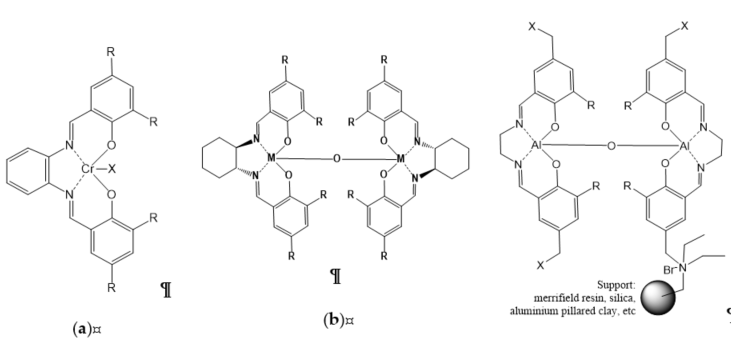
Figure 6. Organometallic salen-based catalysts: (a) homogeneous monometallic salen complexes; (b) homogeneous bimetallic salen complex; (c) heterogeneous bimetallic salen complex supported by various supports.
3.1.1. Organocatalysts
The characteristics of organocatalysts are readily available, non-toxic, affordable, and inert towards air. Thus, organocatalysts are easy to handle and have recently attracted considerable attention and interest in this field as an alternative to the metal-based catalyst. Several types of organocatalysts including, but not limited to, organic salts and ionic liquids, amines, organic bases, and deep eutectic solvents are of major interest among researchers.
Quarternary ammonium halides, one of the organic salts, were among the first active catalysts for the cyclic carbonates synthesis [27]. Tetrabutylammonium bromide (TBAB) effectively catalyzed ethylene oxide cycloaddition to ethylene carbonate with 97% yield, but at a harsh condition of 200 °C and initial CO2 pressure of 34 bar [27]. Other organic salts such as phosphonium salt and imidazolium salt have been later introduced as effective organocatalysts.[28]
Ionic liquid as a catalyst such as imidazolium salt-based ionic liquids (ILs), namely, 1-n-butyl-3-methylimidazolium tetrafluoroborate, exhibited 100% propylene oxide conversion with almost 100% selectivity of propylene carbonate at 110 °C and CO2 initial pressure of 40 bar. Good recyclability can be obtained with a slight decrease in conversion and selectivity after 5 cycle runs[29]. The ILs are defined as salts that melt at 100 °C or less. Organic salts and ILs can be classified and differentiated by their cation structures. The mechanism of these organocatalysts is intrinsically based on the nucleophilic attack of the anion and the stabilization by interacting with the cation [30]. Because the reaction occurs in the liquid phase, the solubility of the catalyst plays an important role. Many types of ILs have been used to catalyze the reactions, including ILs based on imidazolium, ammonium, phosphonium, and pyridinium salts. For example, Wang et al. studied the dual functional catalytic system by incorporating pyridine alcohol with tetrabutylammonium iodide (TBAI). The results of the binary catalytic showed increase of yield of the different usage of halides ions in the order of Clˉ < Brˉ < Iˉ with yield of 52%, 67%, and 92%, respectively (Table 1). This is probably because chloride ion has stronger hydrogen bond interactions with the alcohol component, when compared to bromide and iodide ions. This study displays conversion of diverse epoxides with high yield, ranging from 85%–97%, for which ECH is the highest yield due to electron withdrawing effect of the substituent, which favors the nucleophilic attack at the carbon atom of the epoxide ring. The catalytic system of 2,6-pyridinedimethanol/TBAI was sustainable and economical because the activity was almost constant after recycling at least six times.
Deep eutectic solvents (DESs) have recently emerged as captivating multifunctional media. DESs possess analogous properties of ILs; however, it has been proposed as a promising alternative to conventional ILs because of easier preaparation and lower cost. DESs are eutectic mixtures of hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) in a certain stoichiometric ratio. Tak et al. [20] studied the reaction of DES by using choline chloride (ChCl) as HBA and various HBDs, these being urea, ethylene glycol, glycerol, and benzoic acid, with a constant mole ratio of ChCl to HBD of 1:2, finding that urea served as the best HBD of SEO conversion among others because of the greater solubility of CO2 in a ChCl/urea DES mixture (0.301 mol CO2/mol DES). With regard to the activation barrier that hindered the formation of bicyclic spiro-cyclic carbonate, the studies investigated the effect of temperature from 40 to 70 °C, significantly influencing the yield of spiro-cyclic carbonate with the increment of 49% to 89% in 5 h. The effect of catalyst loading of ChCl/urea (100–300 mg) was observed, and 300 mg achieved the highest yield (98%) in a shorter reaction period. The effect of substitutions of benzyl (Bn) attached to nitrogen (Figure 3i) with other substrate (i.e., methyl, allyl, and methylbenzyl) provided excellent yields (95%–98%); in comparison, the substitutions of aromatic ring-derived substrates (Figure 3i); R=H, F, Cl, benzyl, etc.) required additional duration (6–8 h) in achieving a comparable yield (67%–90%), which was somewhat expected due to the steric hindrance of the substituted substrate, and the author suggested that providing higher pressure and temperature may achieve better conversion. The recyclability of ChCl/urea DES displayed consistent yield (≈98%) in four runs.
3.1.2. Metal Salt
The most commonly used metal salts are those based on alkali metal salts (also called alkali-metal halides). Alkali metal salts of sodium, potassium, or lithium, which are abundant, low-cost, and non-toxic can be employed to catalyze the CO2/epoxide coupling. For instance, many researchers reported the production of ethylene carbonate under high temperature and pressure. For example, Dow Chemical and Shell patented KI as a catalyst for the ethylene carbonate production from CO2 and ethylene oxide under harsh conditions, around T = 190 °C and P = 13 bar . For propylene carbonate production, 99% of propylene oxide conversion can be achieved at 120 °C and 30 bar within 5 h when the reaction is catalyzed by KI, whereas only 27% and 3% conversion occurs in the cases of KBr and KCl catalysts, respectively [31]. However, the activity of alkali metal salts is relatively low, and therefore a co-catalyst is commonly necessary [32][33].
3.1.3. Metallic Salen Complex
Monometallic Salen Complex
Homogeneous metal–salen and affine complexes (salphen, salophen, salalen, etc.) have been well developed over the last two decades as salen ligands that can coordinate with many transition metals, and have been widely studied for cycloaddition reaction . Salen complexes are readily available, and can be finely tuned and prepared in enantiomerically pure form, thus enabling the creation of an asymmetric environment around the active metal site . These metal complex catalysts have been used in many organic reactions, targeting cycloaddition reaction of CO2. Castro-Osma et al. found an interesting finding of Cr(III) salen complexes with the use of the salophen ligand and tetrabutylammonium bromide (TBAB) that could catalyze CO2 coupling under ambient condition (Figure 6). Regarding the monometallic salen complex (Figure 6a; R=tert-butyl, X=Cl) with TBAB as a co-catalyst, the cycloaddition of CO2 with styrene oxide gave 100% conversion with selectivity higher than 99% under ambient condition for 24 h. Modifying the salen complex (Figure 6a; R=tert-butyl, X=Cl) by replacing the tert-butyl substituents with methoxy groups (Figure 6a; R=MeO, X=Br), results in higher rate of reaction, verifying the fact that replacing substituent with electron-donating groups (MeO) could produce more active catalysts.
Bimetallic Salen Complexes
The development of bimetallic salen complex with TBAB as co-catalyst was the earliest catalytic system capable of catalyzing the insertion of CO2 into terminal epoxides at ambient pressure and temperature [34]. On the basis of Wang et al. , the calculation using density functional theory (DFT) proved bimetallic (salen-Co) complexes were one of the most efficient catalysts, with an activation energy of merely 9.94 kcal/mol, indicating that the reaction could occur under ambient conditions. The experimentally produced yield of 75.8% with salen-Co catalyst was the highest yield attained when compared with salen-Al and salen-Zn at ambient conditions (Figure 6b).
3.1.4. Metallic Non-Salen-Based Complexes
Monometallic Non-Salen-Based Complexes
Due to a unique framework of porphyrin, coordination with metal referred to as “metalloporphyrin” is highly versatile and allows tunable electronic properties of the metal center, which can enhance catalytic activity and exhibit good thermal stability . It is one of the most studied organometallic complexes besides salen-based complexes. For example, Maeda et al. studied functionalized Zn(II) TPP (tetraphenylporphyrin) with eight quaternary ammonium bromides at the ortho, meta, or para positions of the meso-phenyl groups (Figure 7a; R=O(CH2)6N+Bu3Br−). The meta-substituted Zn(II) complexes showed very high activity. At 20 °C, they reported a cyclic carbonate yield of 82% with a TON (turnover number) of 1640 in 48 h under atmospheric pressure of CO2. A very high TON of 240,000 can be achieved by increasing reaction temperature to 120 °C and initial CO2 pressure to 17 bar. In a mechanistic approach, DFT (density functional theory) calculation was employed to reveal the origin of merit of the meta-substituted catalyst.
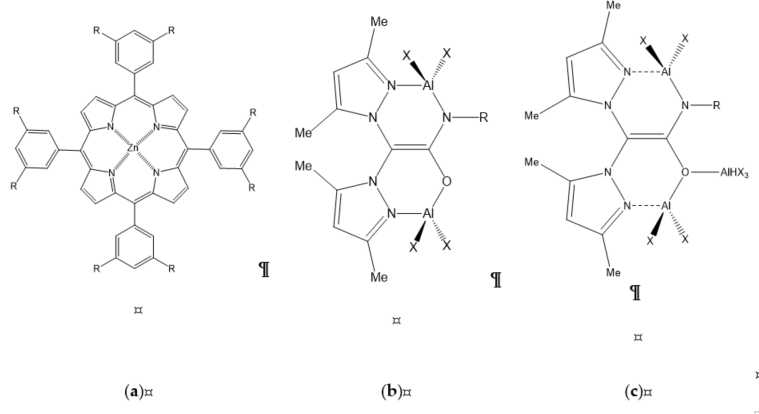
Figure 7. Non-salen-based complexes: (a) metalloporphyrin-based complex; (b) binuclear aluminum scorpionate; (c) trinuclear aluminum scorpionate.
De et al. reported the synthesis of nonsymmetric aza-oxa cryptand derivatized with L-proline. The trinuclear Co(II) complex {[Co3(L)2(NCS)6]·(15CH3CN) (5acetone)(6H2O)} can be formed by reacting the cryptand and Co(II) perchlorate in the presence of KSCN. With TBAB as a co-catalyst, all different epoxides could be converted to cyclic carbonates with 100% selectivity at 20 °C under atmospheric pressure, however, with low to moderate yields of 32%–49% within 12–24 h of reaction time.
Bimetallic Non-Salen Complexes
Castro-Osma et al. studied bimetallic alluminium scorpionate as non-salen complexes (Figure 7b) and other organometallic complexes. Bimetallic aluminium scorpionate (Figure 7b; R=CH(Ph)Me and X=Et) shows the added benefit of using a bimetallic complex that is very high active over monometallic complex catalysts. However changing this from a large alkyl group (R=CH(Ph)Me; X=Et) to a simple aryl group (R=Ph; X=Me) resulted in a significant decrease in catalytic activity, which is also lower than the activity of the monometallic complex. This is critically due to the nature of the subtituted group attached to the enamine nitrogen atom.
Trimetallic Non-Salen Complexes
In the same study, Castro-Osma et al. also synthesized trimetallic aluminium scorpionate (Figure 7c) by adding trialkylaluminium to the oxygen atom of the previous bimetallic complex. On the basis of Table 1, all trimetallics provide complete conversion of SO with 97.3% yield of styrene carbonate, except R=(S)-CH(Ph)Me; X=Me given 92% conversion and 90.2% yield at 10 bar and room temperature for 24 h. Among trimetallic non-salen complexes, only R=Ph; X=Me can maintain the highest catalytic activity of 97.3% yield of styrene carbonate when the CO2 opearting pressure decreases to 1 bar.
Another example is trimetallic amine-bis(benzotriazole phenolate) complexes with Ni(II), Co(II), and Zn (II) metal centers . Trimetallic-nickel, trimetallic-cobalt, and trimetallic-zinc achieved conversion of 43%, 67%, and 91%. All the catalysts displayed the same trend of catalytic systems, for which TBAB gave better conversion as a co-catalyst than TBAI. A reasonable explanation for TBAB as a better co-catalyst for the cycloaddition of CO2 with CHO might be attributed to the balance between nucleophilicity and leaving ability of the bromide anion.
3.2. Heterogeneous Catalyst
Various heterogeneous catalysts have been developed for the past few decades. The high demand for heterogeneous catalysts is due to the simple separation process . In addition, the advantages of heterogeneous catalyst employment is their high recyclability and recovery of the product and catalyst . However, due to their relatively low catalytic activity, most heterogeneous catalytic systems must be applied under the conditions of high temperature and high pressure or in the presence of a solvent and/or co-catalyst.
3.2.1. Supported Catalyst
A wide range of heterogeneous catalysts have been created, with the active sites being immobilized on various types of support, for example, ionic liquid grafted on polymer matrix [35][36], imidazolium derivatives functionalized on MCM-41 (Mobil Composition of Matter No. 41) silica-based materials [37], or triazolium-based ionic liquids on SBA-15 (Santa Barbara Amorphous-15) [38]. Normally, the covelent immobilization/functionalization is robust and can prevent the leaching of active species from the support.
Supported Ionic Liquid
Han et al. [39] synthesized highly cross-linked poly(N-vinylimidazole-co-divinylbenzene) (PVIm) beads with grafted ILs. By varying molar ratio of divinylbenzene to N-vinylimidazole, various pore sizes could be obtained and the highest IL immobilization of 0.92 mmol/g could be achieved. This study used several alkyl halides that covalently anchored on the PVIm surfaces to obtain variation of ILs-grafted porous polymer beads (PVIm2-RX). However, the author extensively studied PVIm grafted with N-butyl bromide (BuBr) by varied reaction parameters. The effect of temperature (90, 100, and 110 °C) on conversion of allyl glycidyl ether (AGE) showed increasing conversion of 49%, 60%, and 65%, respectively (Table 2). For the effect of pressures on catalytic activity at pressures of 8.6, 13.4, 16.2, and 18.2 bar, AGE conversion increased to 65%, 855, 94%, and 98%, respectively. PVIm-BuBr exhibited a good reusability with almost constant yield for five consecutive runs.
Table 2. Supported catalysts for cycloaddition of CO2 to epoxides.[39][40][41][42]
|
Epoxide |
Catalyst |
Cocatalyst |
T (°C) |
P (bar) |
Time (h) |
Yield (%) |
Conv. (%) |
Reusability |
Ref. |
|
AGE |
PVIm2-BuBr |
- |
90 |
8.6 |
6 |
48.02 |
49 |
5 times, drop slightly after fourth run |
[39] |
|
- |
100 |
8.6 |
6 |
58.8 |
60 |
||||
|
- |
110 |
8.6 |
6 |
64.35 |
65 |
||||
|
- |
110 |
13.4 |
6 |
84.15 |
85 |
||||
|
- |
110 |
16.2 |
6 |
93.06 |
94 |
||||
|
- |
110 |
18.2 |
6 |
97.02 |
98 |
||||
|
SO |
Bimetallic salen-merrifield resin (single TBAB) |
- |
26 |
1 |
20 |
100 |
- |
100%, 94%, 74%, 70% |
[40] |
|
Bimetallic salen-merrifield resin (four TBAB) |
- |
26 |
1 |
20 |
79 |
- |
79%, 71%, 67%, 64% |
||
|
Bimetallic salen-silica supported(R=t-butyl) |
- |
26 |
1 |
24 |
86 |
- |
- |
[41] |
|
|
Bimetallic salen-aluminium pillared clay (R=t-butyl) |
- |
26 |
1 |
24 |
21 |
- |
|||
|
ECH |
MgFeAl-LDH |
TBAB |
25 |
5 |
48 |
92.83 |
96.3 |
- |
[42] |
|
TBAB |
50 |
1 |
7 |
74.28 |
75.8 |
- |
|||
|
TBAB |
50 |
5 |
7 |
96.04 |
98.0 |
- |
|||
|
- |
50 |
5 |
7 |
7.39 |
7.6 |
- |
|||
|
- |
- |
50 |
5 |
7 |
16.6 |
17.0 |
- |
Supported Bimetal–Organic Salen Complexes
Although homogeneous catalysts such as Co(III) and Al(III) salen complexes show an effective catalytic system, it displays poor separation and low recyclability . Thus, North et al. attempted to immobilize bimetallic salen catalysts that can undergo reaction under mild conditions, deeming them as potential catalysts that can be utilized for continuous flow system as packed-bed reactor. This advancement of heterogeneous of bimetal–organic complexes catalysts enables the recovery of catalysts as an essential approach due their relatively high molecular weight and high production costs. This work utilized the salen complexes that are able to operate under ambient conditions by immobilization on Merrifield resin support via a pendant ammonium moiety (tetraalkylammonium bromide). The recyclability of the catalytic system of bimetallic salen (R1=t-butyl, R2=C2H8, X=+NEt2) (Figure 6c), which consists of a single pendant ammonium moiety showing conversion SO in yields of 100%, 94%, and 70% upon two runs. The bimetallic salen (R1=t-butyl, R2=C2H8, X=+NEt2Bn −Br) (Figure 6c) with four quaternary ammonium moieties provided conversion of styrene oxide in four successive runs with yields of 79%, 73%, 66%, and 60%. In the continuation of their study, North et al. [41] attempted to change the Merrifield resin with a few types of supports, such as amorphous silica (Figure 6c), due to the excellent results gained using Merrifield resin-supported complexes. However, the silica-supported and aluminium-pillared clay only provided 86% and 21% yields of cyclic carbonate, respectively. In this study, the continuous flow reactor also operated as a silica-supported thermally stable reactor. The catalyst bimetallic salen (R1=t-butyl, R2=C2H8, X=+NEt2) supported on silica (Figure 6c) at 100 °C showed slow deactivation over the first 8 days, for which it retained 50% conversion, but the restoration by treatment with benzyl bromide provided original activity of the catalyst.
Trimetallic Layered Double Hydroxide (LDH)
Zhang et al.[42] synthesized trimetallic MgFeAl-LDH from industrial solid wastes of red mud and ferronickel slag. The acid-extracted solutions of solid waste and exfoliation of the LDH by washing with acetone resulted in high surface area of MgFeAl-LDH (319 m2/g) and high CO2 uptake of 70.2 mg/g at room temperature and atmospheric pressure. The MgFeAL-LDH with TBAB as co-catalyst was tested for solvent-free CO2 cycloaddition under different pressures. ECH conversion increased from 75.8% to 98% with high selectivity (≈98%) by increasing pressure from 1 to 5 bar. The paper also studied the effect of conversion toward various epoxides (i.e., ECH, PO, SO, CHO, and AGE) at 50 °C and 5 bar after 7 h. The results showed significant conversion and selectivity for all epoxides, for which epichlorohydrin attained the highest conversion of 98.0% and 98.2% selectivity. To verify the synergistic effect between Lewis acid sites of MgFeAl-LDH and the bromine nucleophile of TBAB, MgFeAl-LDH and TBAB were tested separately, giving the ECH conversions of 7.6% and 17.0%, respectively. The MgFeAl-LDH showed high recyclability, which provided stable ECH conversion no less than 96% and selectivity around 97% in five consecutive runs.
3.2.2. Metal–Organic Framework
Metal–organic framework (MOF), a combination between metal ions or clusters and interconnected organic ligands, is expected to be a good candidate of heterogeneous catalysts [43]. The distinctive features of adjustable structures, large surface area, and coordinated pore surface portray a vital role in yielding high CO2 sequestration and its effective conversion to valuable products [44,45]. Table 3 summarizes operational conditions for the synthesis of cyclic carbonates using MOFs.
Table 3. Metal organic framework (MOF) catalysts for cycloaddition of CO2 to epoxides.[43][44][45]
|
MOF |
Active Site |
T (°C) |
P (bar) |
Time (h) |
Epoxide |
Yield (%) |
Conv. (%) |
TOF |
Reusability |
Ref. |
|
In2(OH)(btc)(Hbtc)0.4(L)0.6·3H2O |
Metal |
RT |
1.01 |
48 |
PO |
77.9 |
- |
7.1 |
5 times (almost constant) |
[1] |
|
BO |
60.1 |
- |
5.4 |
|||||||
|
SO |
31.6 |
- |
2.9 |
|||||||
|
{[Co(BDC)(L)]·2H2O·xG}n |
Metal and pyridine |
RT |
1.01 |
48 |
SEO |
84.15 |
85 |
- |
5 times (almost constant) |
[44] |
|
40 |
1.01 |
24 |
98.01 |
99 |
- |
|||||
|
{Cu2[(C20H12N2O2)(COO)4]}n |
Metal and acylamide |
RT |
1.01 |
48 |
PO |
96 |
- |
- |
(96%, 96%, 95%, 95%, 95%) |
[43] |
|
BO |
85 |
- |
- |
|||||||
|
ECH |
88 |
- |
- |
|||||||
|
Octene oxide |
10 |
- |
- |
|||||||
|
{Cu4[(C57H32N12)(COO)8]}n |
Metal and triazole |
RT |
1.01 |
48 |
PO |
96 |
- |
200 |
(96%, 96%, 96%, 95%, 95%) |
[45] |
|
BO |
83 |
- |
172.9 |
|||||||
|
ECH |
85 |
- |
177 |
Liu et al. [1] synthesized In2(OH)(btc)(Hbtc)0.4(L)0.6·3H2O (Figure 8a) with TBAB as cocatalyst. The catalyst retained excellent recyclability with no significant change within five cycles of reaction. The catalyst also showed high selectivity toward size and shape of the substrate. PO showed the highest (77.9%) reactivity among substrates, which was likely caused by sterically hindrance for diffusion onto the pore of MOF.
There are several researchers who have demonstrated that incorporation of accessible nitrogen donor sites, such as amine, imidazole, pyridine, tetrazole, or triazole into MOFs can drastically influence the CO2 sorption capacity and selectivity on account of dipole–quadrupole interactions between the CO2 molecule and the accessible nitrogen site[46][47][48][49][50].
Parmar et al. synthesized [Co(BDC)(L)·2H2O]·xG}n or CoMOF-2 (Figure 8b) with nitrogen-rich pyridine as basic site. They demonstrated an excellent catalytic activity of CoMOF-2 and KI as binary catalyst in epoxide−CO2 cycloaddition. SEO conversion of 85% was obtained at room temperature for 48 h. In addition, 99% conversions were achieved within 24 h, with increasing temperature to 40 °C. The recyclability remained constant with 99% conversion and yield through five successive cycles.
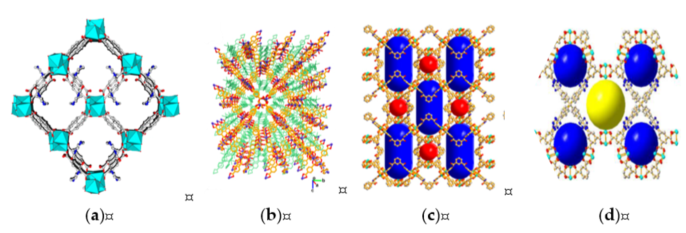
Figure 8. The 3D structure of MOFs: (a) In2(OH)(btc)(Hbtc)0.4(L)0.6·3H2O (reprinted with permission from Liu, L.; Wang, S.M.; Han, Z.B.; Ding, M.; Yuan, D.Q.; Jiang, H.L. Exceptionally Robust In-Based Metal-Organic Framework for Highly Efficient Carbon Dioxide Capture and Conversion. Inorg. Chem. 2016, 55, 3558–3565. Copyright (2016) American Chemical Society.); (b) {[Co(BDC)(L)]·2H2O·xG}n (CoMOF-2) (reprinted with permission from Parmar, B.; Patel, P.; Pillai, R.S.; Tak, R.K.; Kureshy, R.I.; Khan, N.H.; Suresh, E. Cycloaddition of CO2 with an Epoxide-Bearing Oxindole Scaffold by a Metal–Organic Framework-Based Heterogeneous Catalyst under Ambient Conditions. Inorg. Chem. 2019, 58, 10084–10096. Copyright (2019) American Chemical Society.); (c) {Cu2[(C20H12N2O2)(COO)4]}n (reprinted with permission from Li, P.Z.; Wang, X.J.; Liu, J.; Phang, H.S.; Li, Y.; Zhao, Y. Highly Effective Carbon Fixation via Catalytic Conversion of CO2 by an Acylamide-Containing Metal-Organic Framework. Chem. Mater. 2017, 29, 9256–9261. Copyright (2017) American Chemical Society.); (d) {Cu4[(C57H32N12)(COO)8]}n (reprinted with permission from Li, P.Z.; Wang, X.J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A Triazole-Containing Metal-Organic Framework as a Highly Effective and Substrate Size-Dependent Catalyst for CO2 Conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. Copyright (2016) American Chemical Society.).
Li et al. synthesized Cu2[(C20H12N2O2)(COO)4] (Figure 8c), which incorporated both accessible nitrogen-rich acylamide groups and exposed Cu metal sites expressing a high CO2 adsorption capability. The results showed 96% for propylene oxide, 85% for butylene oxide, 88% for epichlorohydrin, and 10% 1-octene oxide (1,2-epoxyoctane) on cycloaddition at 1.01 bar of CO2 and room temperature, which demonstrated high selectivity toward size and shape of substrate. The catalyst displayed slight decrease of conversion of propylene oxide after second run within five cycles.
Li et al. utilized a highly porous triazole-based MOF, Cu4[(C57H32N12)(COO)8] (Figure 8d), which showed high activity towards size- and shape-selective synthesis of carbonates. MOF incorporating both unsaturated metal sites and accessible nitrogen-rich triazole units exhibited a high affinity towards CO2. The MOF exhibited high catalytic selectivity to small epoxide substrate—96% for propylene oxide, 83% for butylene oxide, and 85% for epichlorohydrin, with a very low yield for large epoxide substrate. The catalyst displayed slight (≈1%) decrease of conversion after the third run within five cycles. On the basis of this observation, MOF is an effective catalyst for CO2 cycloaddition reaction, specifically in terms of recyclability and selectivity.
References
- Liu, L.; Wang, S.M.; Han, Z.B.; Ding, M.; Yuan, D.Q.; Jiang, H.L. Exceptionally Robust In-Based Metal-Organic Framework for Highly Efficient Carbon Dioxide Capture and Conversion. Inorg. Chem. 2016, 55, 3558–3565.
- Bals, C.; Bellmann, E.; Bode, A.; Edenhofer, O.; Fischedick, M.; Gaertner, L.-E.; Gerling, P.; Helseth, J.M.; Kühn, M.; Liebscher, A. CCU and CCS–Building Blocks for Climate Protection in Industry. Analysis, Options and Recommendations; Utzverlag GmbH: München, Germany, 2019.
- Zhang, Z.; Pan, S.Y.; Li, H.; Cai, J.; Olabi, A.G.; Anthony, E.J.; Manovic, V. Recent advances in carbon dioxide utilization. Renew. Sustain. Energy Rev. 2020, 125, 109799.
- Liang, S.; Liu, H.; Jiang, T.; Song, J.; Yang, G.; Han, B. Highly efficient synthesis of cyclic carbonates from CO2 and epoxides over cellulose/KI. Chem. Commun. 2011, 47, 2131–2133.
- Ng, C.K.; Toh, R.W.; Lin, T.T.; Luo, H.K.; Hor, T.S.A.; Wu, J. Metal-salen molecular cages as efficient and recyclable heterogeneous catalysts for cycloaddition of CO2 with epoxides under ambient conditions. Chem. Sci. 2019, 10, 1549–1554.
- Lu, B.B.; Yang, J.; Liu, Y.Y.; Ma, J.F. A Polyoxovanadate-Resorcin[4]arene-Based Porous Metal-Organic Framework as an Efficient Multifunctional Catalyst for the Cycloaddition of CO2 with Epoxides and the Selective Oxidation of Sulfides. Inorg. Chem. 2017, 56, 11710–11720.
- Styring, P.; Jansen, D.; de Coninck, H.; Reith, H.; Armstrong, K. Carbon Capture and Utilisation in the Green Economy; Centre for Low Carbon Futures: New York, NY, USA, 2011.
- Aresta, M. Carbon dioxide utilization: Chemical, biological and technological applications. In Greenhouse Gases: Mitigation and Utilization, Proceedings of the CHEMRAWN-XVII and ICCDU-IX Conference, Kingston, ON, Canada, 8–12 July 2007; Queen's University: Kingston, ON, Canada, 2009; pp. 123–149.
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 1–15.
- Luo, J.; Preciado, S.; Xie, P.; Larrosa, I. Carboxylation of Phenols with CO2 at Atmospheric Pressure. Chem. Eur. J. 2016, 22, 6798–6802.
- Wendling, T.; Risto, E.; Krause, T.; Gooßen, L.J. Salt-Free Strategy for the Insertion of CO2 into C−H Bonds: Catalytic Hydroxymethylation of Alkynes. Chem. Eur. J. 2018, 24, 6019–6024.
- Zou, B.; Hu, C. Synthesis of Cyclic Carbonates from Alkenyl and Alkynyl Substrates. Chin. J. Chem. 2017, 35, 541–550.
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246.
- Wang, L.; Zhang, G.; Kodama, K.; Hirose, T. An efficient metal- and solvent-free organocatalytic system for chemical fixation of CO2 into cyclic carbonates under mild conditions. Green Chem. 2016, 18, 1229–1233.
- Jadhav, A.H.; Thorat, G.M.; Lee, K.; Lim, A.C.; Kang, H.; Seo, J.G. Effect of anion type of imidazolium based polymer supported ionic liquids on the solvent free synthesis of cycloaddition of CO2 into epoxide. Catal. Today 2016, 265, 56–67.
- North, M. Synthesis of Cyclic Carbonates from Carbon Dioxide and Epoxides; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 9780444538826.
- Bobadilla, L.F.; Lima, S.; Urakawa, A. Cycloaddition of CO2 and Epoxides over Reusable Solid Catalysts. Adv. Catal. Mater. 2015, 271–312, doi:10.1002/9781118998939.ch8.
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450.
- Gruttadauria, M.; Giacalone, F. Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011; ISBN 9780470641361.
- Tak, R.K.; Patel, P.; Subramanian, S.; Kureshy, R.I.; Khan, N.U.H. Cycloaddition Reaction of Spiro-Epoxy Oxindole with CO2 at Atmospheric Pressure Using Deep Eutectic Solvent. ACS Sustain. Chem. Eng. 2018, 6, 11200–11205.
- Castro-Osma, J.A.; Lamb, K.J.; North, M. Cr(salophen) Complex Catalyzed Cyclic Carbonate Synthesis at Ambient Temperature and Pressure. ACS Catal. 2016, 6, 5012–5025.
- Wang, T.T.; Xie, Y.; Deng, W.Q. Reaction mechanism of epoxide cycloaddition to CO2 catalyzed by salen-M (M = Co, Al, Zn). J. Phys. Chem. A 2014, 118, 9239–9243.
- Maeda, C.; Shimonishi, J.; Miyazaki, R.; Hasegawa, J.Y.; Ema, T. Highly Active and Robust Metalloporphyrin Catalysts for the Synthesis of Cyclic Carbonates from a Broad Range of Epoxides and Carbon Dioxide. Chem. Eur. J. 2016, 22, 6556–6563.
- De, D.; Bhattacharyya, A.; Bharadwaj, P.K. Enantioselective Aldol Reactions in Water by a Proline-Derived Cryptand and Fixation of CO2 by Its Exocyclic Co(II) Complex. Inorg. Chem. 2017, 56, 11443–11449.
- Castro-Osma, J.A.; Alonso-Moreno, C.; Lara-Sánchez, A.; Martínez, J.; North, M.; Otero, A. Synthesis of cyclic carbonates catalysed by aluminium heteroscorpionate complexes. Catal. Sci. Technol. 2014, 4, 1674–1684.
- Li, C.Y.; Su, Y.C.; Lin, C.H.; Huang, H.Y.; Tsai, C.Y.; Lee, T.Y.; Ko, B.T. Synthesis and characterization of trimetallic cobalt, zinc and nickel complexes containing amine-bis(benzotriazole phenolate) ligands: Efficient catalysts for coupling of carbon dioxide with epoxides. Dalt. Trans. 2017, 46, 15399–15406.
- Lichenwalter, M.; Cooper, J. Catalytic Process for Producing Alkylene Carbonates. U.S. Patent Application No. 2,773,070, 4 Dec 1956.
- Büttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2017; Volume 375, pp. 89–144.
- Peng, J.; Deng, Y. Cycloaddition of carbon dioxide to propylene oxide catalyzed by ionic liquids. New J. Chem. 2001, 25, 639–641.
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 2017, 7, 2651–2684.
- Zhou, X.; Yang, X.; Yao, J.; Wang, G. Synthesis of propylene carbonate from CO2 and propylene oxide with KI and inorganic ammonium salts as catalyst. Acta Chim. Sin. 2010, 68, 870–874.
- Kihara, N.; Hara, N.; Endo, T. Catalytic Activity of Various Salts in the Reaction of 2,3-Epoxypropyl Phenyl Ether and Carbon Dioxide under Atmospheric Pressure. J. Org. Chem. 1993, 58, 6198–6202.
- Huang, J.W.; Shi, M. Chemical fixation of carbon dioxide by NaI/PPh3/PhOH. J. Org. Chem. 2003, 68, 6705–6709.
- North, M.; Quek, S.C.Z.; Pridmore, N.E.; Whitwood, A.C.; Wu, X. Aluminum(salen) complexes as catalysts for the kinetic resolution of terminal epoxides via CO2 coupling. ACS Catal. 2015, 5, 3398–3402.
- Ghazali-Esfahani, S.; Song, H.; Pǎunescu, E.; Bobbink, F.D.; Liu, H.; Fei, Z.; Laurenczy, G.; Bagherzadeh, M.; Yan, N.; Dyson, P.J. Cycloaddition of CO2 to epoxides catalyzed by imidazolium-based polymeric ionic liquids. Green Chem. 2013, 15, 1584–1589.
- Xie, Y.; Zhang, Z.; Jiang, T.; He, J.; Han, B.; Wu, T.; Ding, K. CO2 cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross-linked polymer matrix. Angew. Chem. Int. Ed. 2007, 46, 7255–7258.
- Udayakumar, S.; Lee, M.K.; Shim, H.L.; Park, S.W.; Park, D.W. Imidazolium derivatives functionalized MCM-41 for catalytic conversion of carbon dioxide to cyclic carbonate. Catal. Commun. 2009, 10, 659–664.
- Cheng, W.; Chen, X.; Sun, J.; Wang, J.; Zhang, S. SBA-15 supported triazolium-based ionic liquids as highly efficient and recyclable catalysts for fixation of CO2 with epoxides. Catal. Today 2013, 200, 117–124.
- Han, L.; Choi, H.J.; Kim, D.K.; Park, S.W.; Liu, B.; Park, D.W. Porous polymer bead-supported ionic liquids for the synthesis of cyclic carbonate from CO2 and epoxide. J. Mol. Catal. A Chem. 2011, 338, 58–64.
- Meléndez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579, doi:10.1039/b900180h.
- Zheng, B.; Bai, J.; Duan, J.; Wojtas, L.; Zaworotko, M.J. Enhanced CO2 binding affinity of a high-uptake rht-type metal-organic framework decorated with acylamide groups. J. Am. Chem. Soc. 2011, 133, 748–751.
- Li, P.Z.; Wang, X.J.; Zhang, K.; Nalaparaju, A.; Zou, R.; Zou, R.; Jiang, J.; Zhao, Y. “Click”-extended nitrogen-rich metal-organic frameworks and their high performance in CO2-selective capture. Chem. Commun. 2014, 50, 4683–4685.
- Zheng, B.; Yang, Z.; Bai, J.; Li, Y.; Li, S. High and selective CO2 capture by two mesoporous acylamide-functionalized rht-type metal-organic frameworks. Chem. Commun. 2012, 48, 7025–7027.
- Diao, K.S.; Wang, F.; Wang, H. jun Ab initio theoretical study of the interactions between CFCs and CO2. J. Mol. Struct. 2009, 913, 195–199.
- Zhang, M.; Yu, Y. Dehydration of ethanol to ethylene. Ind. Eng. Chem. Res. 2013, 52, 9505–9514.
- Mendieta, C.M.; Vallejos, M.E.; Felissia, F.E.; Chinga-Carrasco, G.; Area, M.C. Review: Bio-polyethylene from Wood Wastes. J. Polym. Environ. 2020, 28, 1–16.
- Broeren, M. Production of Bio-Ethylene; IEA-ETSAP and IRENA: 2013; pp. 1–20.
- Zacharopoulou, V.; Vasiliadou, E.S.; Lemonidou, A.A. One-step propylene formation from bio-glycerol over molybdena-based catalysts. Green Chem. 2015, 17, 903–912.
- Jong, E.D.; Higson, A.; Walsh, P.; Wellisch, M. Task 42 Biobased Chemicals—Value Added Products from Biorefineries. A Rep. Prep. IEA Bioenergy-Task; 2011, Volume 42.
- Resul, M.F.M.G.; López Fernández, A.M.; Rehman, A.; Harvey, A.P. Development of a selective, solvent-free epoxidation of limonene using hydrogen peroxide and a tungsten-based catalyst. React. Chem. Eng. 2018, 3, 747–756.


