 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Judith Blaine | + 5666 word(s) | 5666 | 2020-08-05 10:03:14 | | | |
| 2 | Camila Xu | -147 word(s) | 5519 | 2020-08-06 03:13:27 | | | | |
| 3 | Camila Xu | Meta information modification | 5519 | 2020-08-06 03:14:08 | | | | |
| 4 | Camila Xu | -65 word(s) | 5454 | 2020-08-13 05:30:37 | | | | |
| 5 | Camila Xu | -1 word(s) | 5453 | 2020-10-27 07:49:45 | | |
Video Upload Options
Proteinuria is one of the hallmarks of kidney disease. Serum proteins such as albumin are prevented from being filtered into the urine by the glomerular filtration barrier of which podocytes are a key part. Podocyte structure and function are dependent on maintenance of the actin cytoskeleton in podocyte foot processes. Foot processes contain two structural and signaling hubs: the slit diaphragm and focal adhesions, both of which maintain foot process integrity and relay signals to and from the podocyte exterior in response to hemodynamic changes. The entry below describes the key components of foot process actin cytoskeleton structure and regulation.
1. Introduction or History
The glomerular filtration barrier, of which podocytes are an essential part, prevents serum proteins from being filtered into the urine. Normal podocyte function depends on maintenance of the actin cytoskeleton in these cells.
Proteinuria is one of the hallmarks of kidney disease. Under normal circumstances, less than 150 mg a day of protein is found in the urine. Leakage of serum proteins into the urine is prevented by the glomerular filtration barrier (GFB), a structure consisting of fenestrated endothelial cells, the glomerular basement membrane (GBM) and podocytes. Podocytes are terminally differentiated cells that form the outer layer of the GFB and are exposed to significant mechanical forces: tensile stress and fluid flow shear stress [1]. Tensile stress is generated by changes in glomerular capillary pressure which stretches podocyte foot processes. Shear stress is generated by the flow of glomerular ultrafiltrate which generates stress on the slit diaphragm, major processes and cell body [2]. Podocytes respond to changes in hemodynamic forces within the glomerulus by modifying the foot process actin cytoskeleton. Maintenance of the actin cytoskeleton and actin cytoskeletal dynamics are thus critical for normal podocyte function and preservation of the integrity of the glomerular filtration barrier. Podocyte actin dynamics are controlled by two structural elements within the podocyte, the slit diaphragm (SD) and focal adhesions (FAs), as well as other mediators such as cytokines and drugs. Actin cytoskeleton dysfunction in podocytes can be caused by both acquired and genetic factors. Below, we review the major structural components that regulate the actin cytoskeleton in podocytes and genetic and acquired factors that can modulate actin cytoskeleton dynamics. Before addressing the actin cytoskeleton in podocytes in particular, we briefly review the mechanisms underlying actin cytoskeleton regulation.
2. Structural Regulation of Actin Dynamics in Podocytes
2.1. Actin Dynamics
Changes in the actin cytoskeleton within a cell are necessary for maintenance of cell shape, cell motility and intracellular transport [3]. To carry out these functions, actin filaments are arranged in a variety of ways. The building blocks of actin filaments are 42 kDa actin monomers [4]. Filaments are extended by nucleation, or the formation of actin dimers and trimers, which is the rate limiting step for filament extension [5]. As soon as actin trimers are formed, they are rapidly added to the growing actin filament. Actin filaments are polar structures with two different ends: a barbed end which is highly dynamic and elongates rapidly and a pointed end which elongates at a rate ten-fold slower than that of the barbed end [6]. At the front of the cell, actin filaments are arranged into branched and cross-linked structures known as lamellipodia. Filopodia are protrusions of aligned actin bundles that extend from the front of the cell allowing for directional movement. Actin-generated forces are important for maintaining cell shape and for cell motility. Pushing forces are generated by actin polymerization, whereas pulling forces are generated by the sliding of actin filaments along the filaments of myosin II [5]. Actin polymerization into filaments is tightly controlled by actin-binding proteins [7].
2.2. Podocyte Structure
Podocytes, a key part of the glomerular filtration barrier, prevent high-molecular-weight proteins from entering the glomerular ultrafiltrate. These cells have a unique structure which has been preserved over hundreds of millions of years [8]. The podocyte cell body essentially floats in the ultrafiltrate and is attached to the glomerular basement membrane (GBM) only by its processes. Podocyte primary processes divide to form secondary and tertiary processes which ultimately end in membrane extensions called foot processes (FPs), which interdigitate with neighboring podocytes [9][10][11] (Figure 1). Recently, a fourth structural compartment has been identified in podocytes—a ridge-like prominence which extends directly from the basal surface of the cell body and the primary processes [12]. Primary processes contain microtubules whereas foot processes contain actin filaments [13]. In several vertebrate species, FPs contain two types of actin cytoskeleton: a central actin bundle consisting of loosely assembled actin filaments arranged along the FP longitudinal axis surrounded by a cortical actin network consisting of actin filaments bound to cortactin [14].
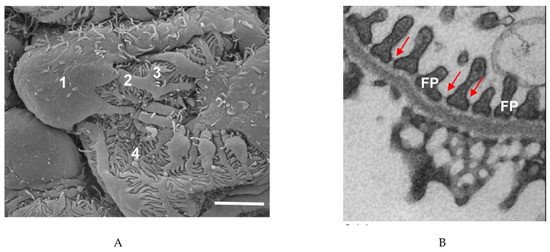
Figure 1. Podocyte structure. (A) Scanning electron microscope image of a podocyte. 1, cell body; 2, major process; 3, secondary process; 4, foot process. Scale bar: 3 µm. (B) Transmission electron microscope image of podocyte foot processes (FPs) and the slit diaphragms between each process (arrows). Scale bar: 1 µm.
Foot processes can be divided into three domains: the apical membrane domain, the slit diaphragm (SD) and the basal membrane domain which is in contact with the glomerular basement membrane [15]. All three domains are physically linked to the FP actin cytoskeleton. Foot processes of neighboring podocytes are connected by slit diaphragms which are multiprotein complexes that act as signaling hubs. FPs also express large macromolecular complexes called focal adhesions which connect the podocyte cytoskeleton to the extracellular matrix [16]. These adhesions not only allow podocytes to adhere to the basement membrane but also relay signals. As can be seen from the structural organization of the podocyte, the actin cytoskeleton is critical to podocyte structure and function.
FPs do not contain contractile actin fibers yet must adapt to changing tensile and shear stresses. Changes in FP shape are likely mediated by a network of myosin IIA-containing contractile fibers which are found within the podocyte cell body and major processes [17]. It has been proposed that the contractile elements in the cell body and major processes generate tension by exerting force on the non-contractile actin filaments in the FPs. This localized tension on integrins induces the assembly of and strengthens focal adhesion complexes which link integrins to the actin cytoskeleton [17]. These forces are also believed to maintain the space between FPs to allow for efficient filtration.
During podocyte injury in vivo, foot processes undergo effacement, becoming flatter and wider [18]. Foot process effacement (FPE) is seen in all glomerular diseases and is particularly noticeable in inflammatory diseases which cause rapidly progressive glomerulonephritis [8]. In the first stage of foot process effacement, slit diaphragms disappear or are displaced from their usual place at the base of the foot process and are replaced by occludens-type junctions [8]. In the second and final stage of FPE, the foot processes retract into primary podocyte processes which results in the cell body adhering to the GBM. During this stage, the actin cytoskeleton within the cell body undergoes extensive rearrangement to form a dense “mat” in close proximity to the GBM. In addition, after injury, myosin IIA redistributes from the major processes and cell body to the basolateral aspects of the cell adjacent to the GBM [17]. In animal models of glomerular diseases such as Masugi nephritis, membranous nephropathy and IgA nephropathy, these changes in the cytoskeleton are accompanied by increased expression of actin, α-actinin and synaptopodin [19][20]. Podocyte injury in vitro results in a more motile phenotype which is thought to correspond to FPE in vivo [8][21]. In culture, injured podocytes also develop an increased number of lamellipodia and filopodia.
Foot processes are dynamic structures and can reassemble after injury as long as the podocyte cell body is attached to the glomerular basement membrane [13]. Actin dynamics in FPs are controlled by two distinct macromolecular protein hubs: the slit diaphragm and focal adhesions (Figure 2).
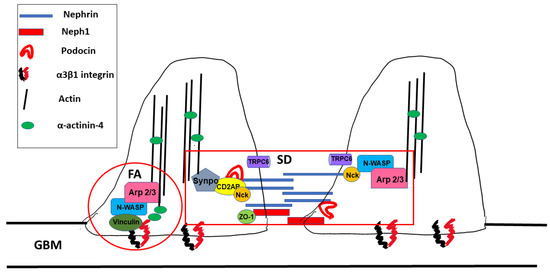
Figure 2. Macromolecular hubs within podocyte foot processes. SD: slit diaphragm, FA: focal adhesion, and GBM: glomerular basement membrane.
2.3. The Slit Diaphragm
The slit diaphragm (SD) is a size-selective barrier between two podocyte foot processes that prevents filtration of large macromolecules into the ultrafiltrate. The SD is also subject to significant shear stress from glomerular filtrate flow [22] (see Section 3.1 below). The SD contains proteins expressed in adherens and tight junctions such as P-cadherin, zonula occludens-1 (ZO-1) and Fat as well as proteins unique to podocytes alone such as nephrin, nephrin-like 1 (neph1) and podocin [23]. During development, the slit diaphragm (SD) initially appears as a cell–cell junction which then matures into a zipper-like structure which connects adjoining podocytes. The slit diaphragm contains many proteins which are involved in maintenance of the actin cytoskeleton and podocyte structure as well as in signaling. After glomerular injury and as chronic kidney disease progresses, SDs are replaced by tight junctions [24][25]. Conceptually, the slit diaphragm can be thought of as being composed of signal receptors (nephrin, neph1 and podocin) which link to signal integrators or adaptors that then link to the actin cytoskeleton (Figure 2). Below, we describe some of the major components of the SD and their role in podocyte actin cytoskeleton dynamics.
2.4. Signal Receptors
2.4.1. Nephrin
Nephrin is a 180 kDa protein encoded by NPHS1. Mutations in nephrin result in congenital nephrotic syndrome of the Finnish type [26]. Nephrin is comprised of a cytoplasmic domain, a short transmembrane domain and an extracellular domain with eight IgG-like motifs and a fibronectin type 3 repeat [27]. Nephrin interacts in a homophilic manner with other nephrin molecules and in a heterophilic manner with neph1 proteins via cis- and trans-interactions (see below) [28][29]. Together, the extracellular domains of nephrin and neph1 form the zipper-like meshwork which creates a sieve-like network with pores with an estimated radius of 12.1 nm each. Nephrin has been shown to be critical for SD formation as without nephrin, the mammalian SD fails to form [30].
2.4.2. Nephrin Signaling
The cytoplasmic portions of both nephrin and neph1 play a major role in signaling and are also important for localizing these proteins to the SD. The cytoplasmic tail of nephrin interacts with podocin (NPHS2, see below) which localizes nephrin to lipid rafts within the SD [31]. Extracellular signals are transmitted to the actin core of the each foot process by tyrosine phosphorylation of nephrin. Tyrosine phosphorylation of nephrin occurs in two distinct domains: group A and group B tyrosines [23]. Phosphorylation of either group A or group B tyrosines regulate cytoskeletal organization. Group A tyrosine phosphorylation also modulates cell survival whereas group B mediates nephrin trafficking. In cultured podocytes, tyrosine phosphorylation of nephrin on group A residues mediates lamellipodia formation whereas group B phosphorylation regulates actin polymer extension. Lamellipodia formation in cultured podocytes is regulated through the p85/PI3K/Akt/Cas/Crk pathway [21][32]. Foot process spreading in vivo in response to podocyte injury, a correlate of lamellipodia formation in vitro, is blocked by podocyte-specific knockout of CT 10 regulator of kinase 1/2 (Crk1/2) [21], underscoring the importance of this pathway in FP actin cytoskeletal dynamics.
Actin extension in podocytes in vitro involves tyrosine phosphorylation and recruitment of non-catalytic region of tyrosine kinase adptor protein (Nck) [33][34]. After nephrin phosphorylation, Nck binds to phospho-nephrin and to neuronal Wiskott–Aldrich syndrome protein (N-WASP) [33][35]. N-WASP then activates the actin-related proteins 2/3 (Arp2/3) complex, a complex of seven proteins including the actin-related proteins Arp2 and Arp3.The Arp2/3 complex is intrinsically inactive because the Arp2 and Arp3 proteins are held apart from each other. Binding of N-WASP to Arp2/3 moves Arp2 and Arp3 closer to each other which promotes de novo actin nucleation and filament branching [36].
In addition to its regulation by tyrosine phosphorylation, the cytoplasmic tail of nephrin also contains a consensus sequence for serine/threonine phosphorylation and can be phosphorylated by protein kinase C alpha (PKCα) [37]. Phosphorylation of nephrin by PKCα results in clathrin and dynamin-mediated endocytosis of nephrin [38], a pathway that is activated in diabetic nephropathy [37][39].
Nephrin signaling is also regulated by tyrosine phosphatases [40]. The Src homology region 2 domain-containing phosphatase-1 (SHP-1) phosphatase which modulates group A tyrosine residues is upregulated in diabetes [41] and another phosphatase, Cl-Ten (Tensin2) modulates podocyte hypertrophy through mammalian target of rapamycin complex 1 (mTORC1) activation [42].
Work from animal models has shown that nephrin tyrosine phosphorylation is required for maintenance of the slit diaphragm. Conversion of all three group B residues in nephrin to phenylalanine which prevents phosphorylation results in mice that rapidly develop foot process effacement and proteinuria after birth [43].
Besides regulation through phosphorylation, nephrin also binds the adaptor protein CD2-associated protein (CD2AP, see below). CD2AP can interact directly with actin [44] as well as cortactin [45] and synaptopodin [46] (see below). This complex provides a direct link between the signal receptor nephrin and the FP actin cytoskeleton.
2.4.3. Neph1
The neph1 protein shares some homology with nephrin but contains only five extracellular IgG-like domains [47]. Besides maintaining slit diaphragm integrity through its interactions with nephrin, neph1 also transduce signals from the podocyte exterior. Tyrosine phosphorylation of neph1 results in recruitment of growth factor receptor bound protein 2 (Grb2) and actin polymerization [48][49].
2.4.4. Podocin
Podocin is a 383 amino acid protein with a hairpin-like structure with both cytoplasmic N- and C-terminal domains [50][51]. Podocin oligomerizes and is found in lipid rafts, specialized microdomains that are enriched in sphingolipid and cholesterol. Assembly of these lipid rafts clusters nephrin at the SD [51]. Podocin is also required to transport nephrin to the SD [52]. Mutations in NPHS2, the gene encoding podocin, are a significant monogenic cause of nephrotic syndrome in children (see below). Besides binding nephrin, the C-terminus of podocin also binds the adaptor protein CD2AP [51]. Podocin also plays a role in formation of tight junctions between neighboring podocytes by associating with and clustering the tight junction proteins coxsackievirus and adenovirus receptor (CAR) and zonula occludens-1 (ZO-1) [53].
2.5. Adaptor Proteins/Signal Integrators
2.5.1. Adaptor Proteins
Adaptor proteins play a critical role in linking nephrin and podocin to the actin cytoskeleton of the foot processes [54]. Adaptor proteins are non-catalytic molecules that contain specific domains which allow for protein–protein interactions. Several adaptor proteins have been identified as necessary for maintenance of the FP actin cytoskeleton. CD2AP binds both nephrin and podocin and links these proteins to actin [55]. Knockout of CD2AP in mice results in massive proteinuria and death by 6–7 weeks of age, indicating that CD2AP is necessary for maintenance of the SD [56]. Nck is another adaptor protein that has been extensively studied. Nck binding to nephrin also plays an important role in SD maintenance (see above). Inducible knockout of Nck in mice leads to a reduction in nephrin tyrosine phosphorylation and decreased actin recruitment to the SD [57]. ZO-1 is another adaptor protein that is important for maintenance of SD integrity. ZO-1 interacts directly with neph1 [58] and this interaction is lost when foot processes efface in response to injury [59].
2.5.2. Rho/Small GTPases
The ras homology (Rho) family of small GTPases (RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1) and cell division control protein 42 homolog (Cdc42)) play a pivotal role in actin dynamics, cell shape and motility. Traditionally, Rac1 and Cdc42 were thought to promote podocyte motility at the leading edge of a migrating cell through formation of lamellipodia (Rac1) and filopodia (Cdc42) whereas RhoA was thought to promote formation of contractile actin and myosin containing stress fibers in the cell body and rear of the cell. In accord with this view, an excess of RhoA activity compared to Rac1/Cdc42 had been shown to result in podocytes with a more stationary phenotype and intact foot processes whereas Rac1 and Cdc42 activation had been shown to promote FP motility and effacement [60][61][62]. However, more recent work has demonstrated that the balance of RhoA to Rac1/Cdc42 activity is central to maintenance of the FP actin cytoskeleton, as both constitutive overexpression and inactivation of RhoA cause FP effacement and proteinuria [63][64]. A variety of factors can modulate RhoA, Rac or Cdc-42 activity in podocytes including calcium influx via transient receptor potential cation channel subfamily C member 6 (TRPC6) [65].
2.5.3. Synaptopodin
Synaptopodin is an actin-associated protein expressed in podocyte foot processes [66]. Synaptopodin interacts with α-actinin and this interaction results in bundling and elongation of actin filaments [46]. Synaptopodin also induce stress fibers in cultured podocytes by blocking the Smurf1-mediated ubiquitination and proteasomal degradation of RhoA, resulting in increased RhoA activity [67]. In vitro, synaptopodin suppresses filopodia formation by binding to IRSp53 and blocking the binding of Cdc42 and Mena to insulin receptor substrate p53 (IRSp53) [68]. Treatment of mice with a Mena inhibitor, FP(4)-Mito, reduces proteinuria in a lipopolysaccharide model of podocyte injury [68].
2.5.4. α-actinin-4
α-actinins are a family of proteins that cross-link actin filaments and serve as scaffolds for the assembly of large protein complexes [69]. α-actinin-4 is highly enriched in podocyte foot processes and mutations in this protein are found in FSGS. In addition, dysregulation of α-actinin-4 occurs early in several other forms of nephrotic syndrome including minimal change disease and idiopathic membranous nephropathy [70][71]. This protein has also been shown to be required for podocyte adhesion to the GBM via interactions with integrins [72]. Loss of α-actinin-4 disrupts interactions between the actin cytoskeleton and integrins, decreasing the overall strength of podocyte attachment to the GBM [16].
2.5.5. TRPC6
Transient receptor potential channel 6 (TRPC6) is a non-selective cation channel that is expressed in podocytes at the SD [73][74]. At the SD, TRPC clusters with several other proteins [74][75][76]. Mutations in TRPC6 are associated with FSGS and upregulation of TRPC6 activity has been seen in several proteinuric kidney diseases including autoimmune glomerulonephritis [77]. TRPC6 localization and activity at the SD is modulated by interactions with both nephrin and podocin [78]. In podocytes, TRPC6 also complexes with RhoA [79]. Overexpression of TRPC6 in podocytes results in derangement of the actin cytoskeleton, retraction of podocyte processes, increased cytosolic calcium, activation of RhoA and downregulation of nephrin and synaptopodin [65]. Channel activation in podocytes in vitro or isolated glomeruli has been shown to be mediated by angiotensin II [80][81], diacylglycerol [80], and reactive oxygen species [82]. TRPC6 has also been shown to interact with calpain 1 and 2. The calpains are a family of calcium-dependent cystine proteases that regulate the actin cytoskeleton and cell motility [83]. TRPC6 binding to calpain 1 and 2 induces cleavage of proteins which mediate podocyte adhesion to the GBM, leading to a more motile phenotype. Conversely, knockdown of TRPC6 decreases calpain-initiated cleavage of the podocyte anchoring proteins talin-1, caldesmon-1 and FAK, resulting in podocytes with increased adhesion, decreased motility and increased actin reorganization [83].
2.6. Genetic Mutations Affecting Components of the Slit Diaphragm
2.6.1. Mutations Affecting Nephrin and Podocin
Given the critical role that nephrin plays in maintenance of the SD, it is not surprising that mutations affecting the nephrin gene, NPHS1, are associated with the severe kidney disease congenital nephrotic syndrome of the Finnish type [26][33][35][84][85][86][87]. Additionally, mutations that alter nephrin trafficking or signaling have also been implicated as causing disease. Examples include mutations in the genes GTPase activating protein and VPS9 domains 1 (GAPVD1) and ankyrin repeat and FYVE domain containing 1 (ANKFY1) which were identified to cause a recessive type of nephrotic syndrome by impairing nephrin trafficking[88].
Podocin, another crucial protein of the slit diaphragm that interacts with nephrin and CD2AP, has also been implicated in human disease. Mutations in NPHS2, which codes for podocin, and LIM homeobox transcription factor 1 Beta (LMX1B), a regulator of podocin expression, have been found to cause nephrotic syndrome [89][90][91][92][93]. Since podocin expression is limited to the glomeruli, NPHS2 mutations typically present without significant extrarenal manifestations [92][93][94]. However LMX1B mutations are causative in the syndromic disease nail–patella syndrome characterized by hypoplastic nails and patella, skeletal deformities and varying degrees of nephropathy [89][90][95]. The underlying pathophysiology caused by mutations affecting podocin relates to impaired nephrin signaling due to the inability of nephrin to associate with lipid rafts as well as podocin’s effects on TRPC6 [52][82][96][97][98].
2.6.2. Mutations Affecting Adaptor Proteins/Signal Integrators
CD2AP is scaffolding protein which links nephrin and podocin with the actin cytoskeleton [56][99][100]. Mutations of CD2AP gene have resulted in nephrotic syndrome and FSGS lesions in humans [56][101][102]. The mechanistic cause for disease is still being studied but is believed to be due to CD2AP’s interactions with F-actin through CAPZ as well as its interactions with Rac1 and synaptopodin [15][55][100][103]. Similar to CD2AP, membrane-associated guanylate kinase 2 (MAGI2) is another scaffolding protein that interacts with the nephrin complex and helps maintain the functional structure of the slit diaphragm [104][105]. Mutations in MAGI2 have been found to cause nephrotic syndrome in mice and humans [106]. MAGI2-associated disease is caused in part due to its interaction with tachykinin 2 (TAC2), which interacts with the Rho GTPase deleted in liver cancer 1 (DLC1), as mentioned above, as well as its interaction with α-actinin-4 and synaptopodin [15][107][108].
As demonstrated in the LMX1B mutations causing kidney disease, diseases can arise if transcriptional regulators of slit diaphragm proteins are mutated. MAF BZIP transcription factor B (MAFB) is an important example of this fact as it is a transcription factor that regulates podocin, nephrin, and CD2AP expression [109]. Two mutations in the MAFB gene have been identified as causative in two different syndromic diseases where proteinuria is a feature, multicentric carpotarsal osteolysis [110] and focal segmental glomerulosclerosis with Duane retraction syndrome (FSGS-DRS) [111].
Synaptodopodin is intriguing from a disease modulation standpoint since it regulates actin activity through its effects on α-actinin-4 and through RhoA activation and Cdc42 inhibition [67]. Synaptopodin has been shown to have anti-proteinuric effects and its absence has been noted in several different disease states [68][112][113][114]. Furthermore, mutations affecting synaptodopodin expression have been noted in patients with proteinuric kidney disease [115]. A significant amount of research is aimed at examining how to modulate synaptopodin for treatment benefit since synaptopodin has TRPC6 modifying properties and part of the effectiveness of the anti-proteinuric effects of cyclosporin A is due to modification of synaptopodin [113][116].
α-actinin-4 is critical for bundling F-actin but also links integrins that adhere to the basement membrane to the cytoskeleton [16][18]. Mutations affecting the ACTN4 gene, which codes for α-actinin-4, clinically present as nephrotic syndrome with FSGS but have a wide age range in which disease manifests [115][117][118]. Notably, different mutations in the ACTN4 genes have demonstrated that the absence of α-actinin-4 as well as mutations causing increased affinity for F-actin, both lead to disease [117][118][119][120].
As outlined, regulation of the cytoskeleton dynamic is maintained by a complex balance through RhoA, Rac1, and Cdc42. Given this complexity and need for a balance between all these pathways, numerous gene mutations associated with this system have been identified as causing diseases. Mutations in Rho GDP dissociation Inhibitor alpha (ARHGDIA) and Rho-GTPase activating protein 24 (ARHGAP24) have both been shown to result in increased Rac1 and Cdc42 activity resulting in disease [121][122][123]. Similarly, mutations in KN motif and ankyrin repeat domains 1/2/4 (KANK1, KANK2, and KANK4) have all been reported to be associated with nephrotic syndrome due to their interactions with proteins that interact with RhoA (KANK 1 interacts with synaptopodin and KANK2 interacts with ARHGDIA) [124]. Mutations in FAT1, intersectin 1 (ITSN1), and ITSN2 genes have all been reported to cause nephrotic syndrome due to their adverse effects on Cdc42 activation resulting in impaired podocyte migration [107][125]. One of the most studied mutations affecting Cdc42 are the mutations in inverted formin 2 (INF2) which codes for a formin protein and has been found to cause Charcot–Marie–Tooth disease as well as FSGS without Charcot–Marie–Tooth disease [126][127][128][129][130][131]. Subsequently, mutations in the INF2 gene have been found to be one of the most common causes in familial nephrotic syndrome [130][131].
DLC1 is another example of a Rho GTPase-activating protein in which mutations cause nephrotic syndrome [107]. Since DLC1 is regulated by cyclic dependent kinase 20 (CDK20) and tensin-2 (TNS2), mutations in TNS2 and cyclin-dependent kinase 20 (CDK20) are also causative in some cases of nephrotic syndrome [107].
Podocalyxin is a cell surface sialomucin expressed by several different cell types including podocytes, vascular endothelium, hematopoietic cells and some subsets of neurons, that is important for podocyte actin cytoskeleton remodeling [132]. Podocalyxin’s C-terminal binding motif (DTHL) interacts with Na+/H+ exchanger regulator proteins (NHERF1 and NHERF2) and actin-binding protein ezrin, and can modify the cytoskeleton through these interactions [132]. When podocalyxin is knocked down, podocytes develop abnormal cell morphology. Knockout of the PODXL gene in mice results in loss of foot processes and loss of the slit diaphragm entirely causing a phenotype of anuric renal failure, omphalocele, and perinatal death [133]. A reported loss-of-function mutation in PODXL has been reported clinically with a very similar phenotype to the one observed in PODXL knockout mice [134]. More recently, podocalyxin has been studied outside the kidney for its role in other diseases like cancer, neurologic and atherosclerotic diseases [132][135][136][137].
TPRC6 plays multiple roles in activating RhoA and Rac1 in response to mechanical stress and influences the function of nephrin, podocin, CD2AP and synaptopodin by regulating intracellular calcium [138]. TPRC6 mutations have also been noted in several cases of congenital FSGS [75][139][140][141]. Interestingly, disease severity caused by TRPC6 mutations seems to be dependent on the degree to which they alter intracellular calcium [138][142]. Most of these mutations are gain-of-function mutations mediated in part through angiotensin II that causes excessive intracellular calcium [77][138][139]. There have been mutations reported that result in a loss of function of TRPC6 which cause disease as well [140]. Remarkably, disease presentation can vary widely in age of onset and severity including some families with members with proteinuric disease and some without disease, despite having the same mutation [143]. As such, it has been postulated that a “second hit” is needed to manifest disease. This theory is supported by a transgenic mouse with a TRPC6 mutation that has no evidence of disease at baseline but developed more severe disease after injury than wild type animals [144]. Since TRPC6 is regulated in part by angiotensin II, a significant amount of research is being done to investigate its role in modulating non-congenital glomerular diseases such as diabetic nephropathy and autoimmune glomerulonephritis [138]. Studies looking at modulating TRPC6 in other disease states have yielded some intriguing results [138], but additional studies are needed to determine when and how to optimally alternate TRPC6 signaling in order to derive clinical benefit since both over and under activation of the channel can be associated with disease.
Phospholipase Cε (PLCε1), a cytoplasmic phospholipase, has been found to be important in podocytes due to its function of regulating internal calcium balance by releasing of calcium from internal stores as well as through activating TRPC6 [65][145]. As mentioned, intracellular calcium affects nephrin and synaptopodin expression in addition to RhoA activation [146][147][148]. Mutations in PLCε1 have been reported to cause SRNS because of its effects on nephrin, synaptopodin, TRPC6 and RhoA [146][149][150].
Glomerular epithelial protien-1 (GLEPP1) is a tyrosine phosphatase expressed on podocyte foot processes and suggested to regulate glomerular pressure and affects nephrin content at the slit diaphragm [151][152]. Decreases in GLEPP-1 have been reported in several diseases including a recessive form of nephrotic kidney disease associated with a mutation on the GLEPP-1 gene, PTPRO [114][151][153][154][155]. Intriguingly, PTPRO knockout mice were reported to be non-proteinuric at baseline but did have significant microscopic abnormalities in podocyte structure and spacing [152].
2.7. Focal Adhesions (FAs)
Focal adhesions are specialized complexes within cells where integrin receptors interact with the extracellular matrix outside of the cell and with the actin cytoskeleton within the cell. In podocytes, FAs link the GBM to the actin cytoskeleton of foot processes (Figure 2) and are under significant tensile and shear stress forces. To withstand these forces, the actin cytoskeleton in FAs flows in a retrograde direction in the vicinity of the podocyte membrane in contact with the GBM [13]. In addition, actin is arranged in linear filaments (stress fibers) in the vicinity of FAs and actin filaments are also cross-linked by myosin II and α-actinin, allowing for increased podocyte adhesion to the GBM via actomyosin contractility [13]. Key components of FAs that link the cell exterior to the FP actin array include integrins and GTPases.
2.7.1. Integrins
Integrins link the cytoskeleton to the extracellular matrix and are composed of α and ß subunits. Integrins are classified as laminin-, collagen- or arginine-glycine-aspartic acid-binding receptors based on their heterodimeric composition which allows for binding to different components of the extracellular matrix. In podocytes, the integrin most critical for maintaining the GFB is the laminin-binding integrin α3β1 [156]. In mice, podocyte-specific knockout of either α3 or β1 integrin results in severe proteinuria and death and in humans mutations in the gene encoding α3 result in severe congenital nephrotic syndrome and a highly disorganized GBM [157][158].
In cultured podocytes, integrin ligation results in tyrosine phosphorylation of nephrin [159]. Clustering of integrins also leads to activation of focal adhesion kinase (FAK) [160]. FAK plays an important role in cell motility and migration including lamellipodia formation and the dynamic assembly or disassembly of focal adhesion complexes by regulating Rho and Rac activity [161][162][163]. Activation of integrin α3β1 also leads to actin polymerization through activation of the Arp2/3 complex, cortactin and the WASP family of proteins [13]. α3β1 integrins are linked to Arp2/3 via vinculin [13]. Podocyte-specific knockout of vinculin results in podocytes with altered FA size, mislocalization of ZO-1 and increased cell migration [164].
While integrin α3β1 is critical for normal podocyte function, podocyte injury leads to activation of the constitutively inactive podocyte integrin αvβ3. Activation of αvβ3 integrin is thought to form a complex between soluble urokinase-type plasminogen activator receptor (suPAR), apolipoprotein 1 (ApoL1) and α3βv integrin which propagates podocyte detachment from the GBM [165]. suPAR-induced activation of this integrin also increases activity of TRPC6 [13].
Integrins are linked to the actin cytoskeleton by several cytoskeletal adaptor proteins including talin1. In mice, loss of talin1 in podocytes results in significant disruption of the actin cytoskeleton [166]. In murine models of glomerular disease, there is increased calpain-induced cleavage of talin1 which also results in disruption of the actin cytoskeleton [166].
2.7.2. GTPases
At FAs, the adaptor protein vinculin links integrins directly with protein complexes involved in actin polymerization including Arp2/3, cortactin and N-WASP [13]. The Rho-A-associated protein kinase ROCK is also involved in stress fiber and focal adhesion formation. ROCK induces formation of stress fibers and FAs by phosphorylating myosin light chain. This promotes actin binding by myosin II and increases foot process contractility [167].
Dynamin is another GTPase which is important for FA formation and function in podocytes. In contrast to Rho, Rac and Cdc42 which are small GTPases, dynamin is a large multidomain protein. Dynamin colocalizes with actin and is involved in growth cone and filopodia formation [168][169][170]. Dynamin can interact directly with actin [171][172] and, after forming dynamin rings, can displace the capping protein gelsolin from the barbed ends of actin filaments promoting actin polymerization [173].
2.8. Genetic Mutations Affecting FA Proteins
Since integrin α3β1is the primary integrin adhering the podocyte to the GBM, mutations in genes ITGA3 and ITGB1 which code for integrin α3 and β1, respectively, have been reported to cause a syndromic disease in humans of nephrotic syndrome, interstitial lung disease, and epidermolysis bullosa [158][174]. Similarly, since CD151 is important for proper insertion of integrin α3β1, mutations in the gene CD151 cause a similar syndromic disease of nephropathy, pretibial epidermolysis bullosa and deafness [175].
Epithelial membrane protein 2 (EMP2) is an interesting protein in that it has been found to perform several functions including cell adhesion, trafficking of GPI proteins into lipid rafts, organization of caveolar regulation, and is involved in vascular endothelial growth factor A- (VEGFA-) mediated angiogenesis [176]. Mutations in the EMP2 gene have been proposed to cause a congenital nephrotic syndrome that was supported by abnormalities noted in cultured human podocytes and zebrafish models with the mutation [177][178]. Interestingly, global and podocyte-specific EMP2 knockout mice do not have proteinuria or abnormal glomerular histology [176]. Given the conflicting animal data and limited number of nephrotic patients with this gene mutated, further research is needed. Disorders of EMP2 may ultimately represent genetic mutations that requires a “second hit” injury before the development of disease.
Remarkably, gene mutations in the integrin linker proteins (paxillin and talin), signaling kinases focal adhesion kinase (FAK), integrin-linked protein kinase (ILK), complex protein PINCH, and kindlin-2 have not been reported to be causative in human kidney disease. This might be explained by the fact that global knockouts in these genes in mice leads to significant embryonic abnormalities that lead to non-viable embryos, supporting the critical nature of these proteins [179][180][181][182][183][184].
2.9. Other Actin Associated Genetic Mutations
Actin by itself is inert and needs appropriate bundling and coupling with myosin to form the contractile apparatus that provides the mechanical force needed for movement. Since actin needs myosin for movement, it is not surprising to learn that mutations affecting myosin can cause diseases which alter the actin cytoskeleton. Most notable in regards to podocytes are mutations in the MYH9 gene, which encodes non-muscle myosin class II isoform A, and MYO1E, which is a membrane-associated class I myosin [185][186].
Mutations in MYH9 are associated with an autosomal dominant pattern of familial nephritis, deafness, and macrothrombocytopenia but has been reported to have significant variation in the degree of renal disease [187]. On the other hand, MYO1E is a recessive mutation associated with FSGS without extrarenal manifestations, likely due to the specialized location and the role MYO1E plays in the podocyte [185].
Without appropriate actin binding, actin and myosin dynamics are impaired, leading to ineffective contractions. Proteins anillin, coded on the ANLN gene, and advillin, coded on AVIL gene, are both F-actin-binding proteins needed for podocyte motility and both have mutations that are associated with kidney disease [188][189]. Outside of their F-actin-binding function, anillin and advillin also interact with CD2AP and PLCε1, respectively, of the slit diaphragm [188], to regulate and coordinate motility while maintaining the filtration barrier [147][189].
References
- Srivastava, T.; Dai, H.; Heruth, D.P.; Alon, U.S.; Garola, R.E.; Zhou, J.; Duncan, R.S.; El-Meanawy, A.; McCarthy, E.T.; Sharma, R.; et al. Mechanotransduction signaling in podocytes from fluid flow shear stress. Am. J. Physiol. Ren. Physiol. 2018, 314, F22–F34. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T.M. Ultrastructure of the actin cytoskeleton. Curr. Opin. Cell Biol. 2018, 54, 1–8. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef]
- Kriz, W.; Shirato, I.; Nagata, M.; LeHir, M.; Lemley, K.V. The podocyte’s response to stress: The enigma of foot process effacement. Am. J. Physiol. Ren. Physiol. 2013, 304, F333–F347. [Google Scholar] [CrossRef]
- Pavenstädt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef]
- Andrews, P.M. Investigations of cytoplasmic contractile and cytoskeletal elements in the kidney glomerulus. Kidney Int. 1981, 20, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Miyaki, T.; Kawasaki, Y.; Kinoshita, M.; Kakuta, S.; Sakai, T. Morphological Processes of Foot Process Effacement in Puromycin Aminonucleoside Nephrosis Revealed by FIB/SEM Tomography. J. Am. Soc. Nephrol. 2019, 30, 96–108. [Google Scholar] [CrossRef]
- Sever, S.; Schiffer, M. Actin dynamics at focal adhesions: A common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Kurihara, H.; Sakai, T. Actin filament organization of foot processes in vertebrate glomerular podocytes. Cell Tissue Res. 2007, 329, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007, 17, 428–437. [Google Scholar] [CrossRef]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, H.Y.; Roth, R.; Jain, S.; Heuser, J.E.; Shaw, A.S.; Miner, J.H. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. Jci Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef]
- Shirato, I. Podocyte process effacement in vivo. Microsc. Res. Tech. 2002, 57, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Shirato, I.; Sakai, T.; Kimura, K.; Tomino, Y.; Kriz, W. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am. J. Pathol. 1996, 148, 1283–1296. [Google Scholar] [PubMed]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017, 91, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. (Lausanne) 2018, 9, 302. [Google Scholar] [CrossRef]
- Succar, L.; Boadle, R.A.; Harris, D.C.; Rangan, G.K. Formation of tight junctions between neighboring podocytes is an early ultrastructural feature in experimental crescentic glomerulonephritis. Int. J. Nephrol. Renov. Dis. 2016, 9, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Anderson, J.M.; Kerjaschki, D.; Farquhar, M.G. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am. J. Pathol. 1992, 141, 805–816. [Google Scholar] [PubMed]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Patrakka, J.; Tryggvason, K. Nephrin—A unique structural and signaling protein of the kidney filter. Trends Mol. Med. 2007, 13, 396–403. [Google Scholar] [CrossRef]
- Barletta, G.M.; Kovari, I.A.; Verma, R.K.; Kerjaschki, D.; Holzman, L.B. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J. Biol. Chem. 2003, 278, 19266–19271. [Google Scholar] [CrossRef]
- Gerke, P.; Huber, T.B.; Sellin, L.; Benzing, T.; Walz, G. Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J. Am. Soc. Nephrol. 2003, 14, 918–926. [Google Scholar] [CrossRef]
- Benzing, T. Signaling at the Slit Diaphragm. J. Am. Soc. Nephrol. 2004, 15, 1382–1391. [Google Scholar] [CrossRef]
- Ruotsalainen, V.; Ljungberg, P.; Wartiovaara, J.; Lenkkeri, U.; Kestilä, M.; Jalanko, H.; Holmberg, C.; Tryggvason, K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7962–7967. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Kovari, I.; Soofi, A.; Nihalani, D.; Patrie, K.; Holzman, L.B. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Investig. 2006, 116, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- New, L.A.; Keyvani Chahi, A.; Jones, N. Direct regulation of nephrin tyrosine phosphorylation by Nck adaptor proteins. J. Biol. Chem. 2013, 288, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Blasutig, I.M.; Eremina, V.; Ruston, J.M.; Bladt, F.; Li, H.; Huang, H.; Larose, L.; Li, S.S.; Takano, T.; et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006, 440, 818–823. [Google Scholar] [CrossRef]
- Mullins, R.D. How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 2000, 12, 91–96. [Google Scholar] [CrossRef]
- Quack, I.; Woznowski, M.; Potthoff, S.A.; Palmer, R.; Königshausen, E.; Sivritas, S.; Schiffer, M.; Stegbauer, J.; Vonend, O.; Rump, L.C.; et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J. Biol. Chem. 2011, 286, 12959–12970. [Google Scholar] [CrossRef]
- Waters, A.M.; Wu, M.Y.; Huang, Y.W.; Liu, G.Y.; Holmyard, D.; Onay, T.; Jones, N.; Egan, S.E.; Robinson, L.A.; Piscione, T.D. Notch promotes dynamin-dependent endocytosis of nephrin. J. Am. Soc. Nephrol. 2012, 23, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Tossidou, I.; Teng, B.; Menne, J.; Shushakova, N.; Park, J.K.; Becker, J.U.; Modde, F.; Leitges, M.; Haller, H.; Schiffer, M. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS ONE 2010, 5, e10185. [Google Scholar] [CrossRef]
- Geraldes, P. Protein phosphatases and podocyte function. Curr. Opin. Nephrol. Hypertens. 2018, 27, 49–55. [Google Scholar] [CrossRef]
- Denhez, B.; Lizotte, F.; Guimond, M.O.; Jones, N.; Takano, T.; Geraldes, P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J. Biol. Chem. 2015, 290, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Koh, A.; Jeong, H.; Kim, E.; Ha, T.S.; Saleem, M.A.; Ryu, S.H. C1-Ten is a PTPase of nephrin, regulating podocyte hypertrophy through mTORC1 activation. Sci. Rep. 2017, 7, 12346. [Google Scholar] [CrossRef] [PubMed]
- New, L.A.; Martin, C.E.; Scott, R.P.; Platt, M.J.; Keyvani Chahi, A.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435. [Google Scholar] [CrossRef]
- Lehtonen, S.; Zhao, F.; Lehtonen, E. CD2-associated protein directly interacts with the actin cytoskeleton. Am. J. Physiol. Ren. Physiol. 2002, 283, F734–F743. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.K.; Winata, S.C.; Lyons, R.J.; Hughes, W.E.; Lehrbach, G.M.; Wasinger, V.; Corthals, G.; Cordwell, S.; Daly, R.J. A Cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J. Biol. Chem. 2003, 278, 21805–21813. [Google Scholar] [CrossRef]
- Asanuma, K.; Kim, K.; Oh, J.; Giardino, L.; Chabanis, S.; Faul, C.; Reiser, J.; Mundel, P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J. Clin. Investig. 2005, 115, 1188–1198. [Google Scholar] [CrossRef]
- Khoshnoodi, J.; Sigmundsson, K.; Ofverstedt, L.-G.; Skoglund, U.; Obrink, B.; Wartiovaara, J.; Tryggvason, K. Nephrin promotes cell-cell adhesion through homophilic interactions. Am. J. Pathol. 2003, 163, 2337–2346. [Google Scholar] [CrossRef]
- Garg, P.; Verma, R.; Nihalani, D.; Johnstone, D.B.; Holzman, L.B. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell. Biol. 2007, 27, 8698–8712. [Google Scholar] [CrossRef]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Hattori, S. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 2008, 283, 9177–9186. [Google Scholar] [CrossRef]
- Roselli, S.; Gribouval, O.; Boute, N.; Sich, M.; Benessy, F.; Attié, T.; Gubler, M.C.; Antignac, C. Podocin localizes in the kidney to the slit diaphragm area. Am. J. Pathol. 2002, 160, 131–139. [Google Scholar] [CrossRef]
- Schwarz, K.; Simons, M.; Reiser, J.; Saleem, M.A.; Faul, C.; Kriz, W.; Shaw, A.S.; Holzman, L.B.; Mundel, P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Investig. 2001, 108, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.B.; Simons, M.; Hartleben, B.; Sernetz, L.; Schmidts, M.; Gundlach, E.; Saleem, M.A.; Walz, G.; Benzing, T. Molecular basis of the functional podocin-nephrin complex: Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 2003, 12, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Shono, A.; Tsukaguchi, H.; Yaoita, E.; Nameta, M.; Kurihara, H.; Qin, X.S.; Yamamoto, T.; Doi, T. Podocin participates in the assembly of tight junctions between foot processes in nephrotic podocytes. J. Am. Soc. Nephrol. 2007, 18, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.S. Roles of adaptor proteins in podocyte biology. World J. Nephrol. 2013, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tossidou, I.; Teng, B.; Worthmann, K.; Müller-Deile, J.; Jobst-Schwan, T.; Kardinal, C.; Schroder, P.; Bolanos-Palmieri, P.; Haller, H.; Willerding, J.; et al. Tyrosine Phosphorylation of CD2AP Affects Stability of the Slit Diaphragm Complex. J. Am. Soc. Nephrol. 2019, 30, 1220–1237. [Google Scholar] [CrossRef] [PubMed]
- Shih, N.Y.; Li, J.; Karpitskii, V.; Nguyen, A.; Dustin, M.L.; Kanagawa, O.; Miner, J.H.; Shaw, A.S. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999, 286, 312–315. [Google Scholar] [CrossRef]
- Jones, N.; New, L.A.; Fortino, M.A.; Eremina, V.; Ruston, J.; Blasutig, I.M.; Aoudjit, L.; Zou, Y.; Liu, X.; Yu, G.L.; et al. Nck proteins maintain the adult glomerular filtration barrier. J. Am. Soc. Nephrol. 2009, 20, 1533–1543. [Google Scholar] [CrossRef]
- Huber, T.B.; Schmidts, M.; Gerke, P.; Schermer, B.; Zahn, A.; Hartleben, B.; Sellin, L.; Walz, G.; Benzing, T. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 2003, 278, 13417–13421. [Google Scholar] [CrossRef]
- Wagner, M.C.; Rhodes, G.; Wang, E.; Pruthi, V.; Arif, E.; Saleem, M.A.; Wean, S.E.; Garg, P.; Verma, R.; Holzman, L.B.; et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 2008, 283, 35579–35589. [Google Scholar] [CrossRef] [PubMed]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef]
- Mundel, P.; Reiser, J. Proteinuria: An enzymatic disease of the podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.D.; Altintas, M.M.; Reiser, J. Podocyte GTPases regulate kidney filter dynamics. Kidney Int. 2012, 81, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Jiang, R.; Aoudjit, L.; Jones, N.; Takano, T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ding, J.; Tsai, H.; Li, L.; Feng, Q.; Miao, J.; Fan, Q. Over-expressing transient receptor potential cation channel 6 in podocytes induces cytoskeleton rearrangement through increases of intracellular Ca2+ and RhoA activation. Exp. Biol. Med. 2011, 236, 184–193. [Google Scholar] [CrossRef]
- Mundel, P.; Gilbert, P.; Kriz, W. Podocytes in glomerulus of rat kidney express a characteristic 44 KD protein. J. Histochem. Cytochem. 1991, 39, 1047–1056. [Google Scholar] [CrossRef]
- Asanuma, K.; Yanagida-Asanuma, E.; Faul, C.; Tomino, Y.; Kim, K.; Mundel, P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell. Biol. 2006, 8, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Bartram, M.P.; Habbig, S.; Pahmeyer, C.; Höhne, M.; Weber, L.T.; Thiele, H.; Altmüller, J.; Kottoor, N.; Wenzel, A.; Krueger, M.; et al. Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum. Mol. Genet. 2016, 25, 1152–1164. [Google Scholar] [CrossRef]
- Goode, N.P.; Shires, M.; Khan, T.N.; Mooney, A.F. Expression of alpha-actinin-4 in acquired human nephrotic syndrome: A quantitative immunoelectron microscopy study. Nephrol. Dial. Transplant. 2004, 19, 844–851. [Google Scholar] [CrossRef]
- Wagrowska-Danilewicz, M.; Stasikowska, O.; Danilewicz, M. Immunoexpression of podocyte-associated proteins in acquired human glomerulopathies with nephrotic syndrome. Pol. J. Pathol. 2006, 57, 17–21. [Google Scholar] [PubMed]
- Dandapani, S.V.; Sugimoto, H.; Matthews, B.D.; Kolb, R.J.; Sinha, S.; Gerszten, R.E.; Zhou, J.; Ingber, D.E.; Kalluri, R.; Pollak, M.R. Alpha-actinin-4 is required for normal podocyte adhesion. J. Biol. Chem. 2007, 282, 467–477. [Google Scholar] [CrossRef]
- Dietrich, A.; Gudermann, T. TRPC6. Handb. Exp. Pharm. 2007. [Google Scholar] [CrossRef]
- Dryer, S.E.; Reiser, J. TRPC6 channels and their binding partners in podocytes: Role in glomerular filtration and pathophysiology. Am. J. Physiol. Ren. Physiol. 2010, 299, F689–F701. [Google Scholar] [CrossRef]
- Reiser, J.; Polu, K.R.; Möller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef]
- Kim, E.Y.; Anderson, M.; Wilson, C.; Hagmann, H.; Benzing, T.; Dryer, S.E. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: Essential role of podocin in formation of this complex. Am. J. Physiol.—Cell Physiol. 2013, 305, C960–C971. [Google Scholar] [CrossRef] [PubMed]
- Dryer, S.E.; Roshanravan, H.; Kim, E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1041–1066. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, K.A.T.; Sonneveld, R.; Bakker-van Bebber, M.; Wetzels, J.F.M.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef]
- Tian, D.; Jacobo, S.M.P.; Billing, D.; Rozkalne, A.; Gage, S.D.; Anagnostou, T.; Pavenstädt, H.; Hsu, H.-H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic Regulation of Actin Dynamics and Cell Motility by TRPC5 and TRPC6 Channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef]
- Anderson, M.; Roshanravan, H.; Khine, J.; Dryer, S.E. Angiotensin II Activation of TRPC6 Channels in Rat Podocytes Requires Generation of Reactive Oxygen Species. J. Cell. Physiol. 2014, 229, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, H.; Dryer, S.E. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: Role of podocin and reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014, 306, F1088–F1097. [Google Scholar] [CrossRef] [PubMed]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.-Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Ohsawa, I.; Ohta, S.; Hattori, S. Phosphorylation of Nephrin Triggers Ca2+ Signaling by Recruitment and Activation of Phospholipase C-{gamma}1. J. Biol. Chem. 2009, 284, 8951–8962. [Google Scholar] [CrossRef]
- Li, S.; Wang, Q.; Wang, Y.; Chen, X.; Wang, Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 2009, 23, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bienaimé, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Grosse, R. Get to grips: Steering local actin dynamics with IQGAPs. Embo Rep. 2007, 8, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Hermle, T.; Schneider, R.; Schapiro, D.; Braun, D.A.; van der Ven, A.T.; Warejko, J.K.; Daga, A.; Widmeier, E.; Nakayama, M.; Jobst-Schwan, T.; et al. GAPVD1 and ANKFY1 Mutations Implicate RAB5 Regulation in Nephrotic Syndrome. J. Am. Soc. Nephrol. 2018, 29, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Claverie-Martin, F.; Trindade, A.; Garcia-Gonzalez, N.C.; Callejon, A.C. Novel missense mutation affecting the LIM-A domain of LMX1B in a family with Nail-Patella syndrome. Intractable Rare Dis. Res. 2019, 8, 14–19. [Google Scholar] [CrossRef]
- Andeen, N.K.; Schleit, J.; Blosser, C.D.; Dorschner, M.O.; Hisama, F.M.; Smith, K.D. LMX1B-Associated Nephropathy With Type III Collagen Deposition in the Glomerular and Tubular Basement Membranes. Am. J. Kidney Dis. 2018, 72, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.; Lennon, R. Basement Membrane Defects in Genetic Kidney Diseases. Front. Pediatr. 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.C.; Niaudet, P.; Antignac, C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Asharam, K.; Bhimma, R.; David, V.A.; Coovadia, H.M.; Qulu, W.P.; Naicker, T.; Gillies, C.E.; Vega-Warner, V.; Johnson, R.C.; Limou, S.; et al. NPHS2 V260E Is a Frequent Cause of Steroid-Resistant Nephrotic Syndrome in Black South African Children. Kidney Int. Rep. 2018, 3, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Baylarov, R.; Senol, O.; Atan, M.; Berdeli, A. NPHS2 gene mutations in azerbaijani children with steroid-resistant nephrotic syndrome. Saudi J. Kidney Dis. Transplant. 2020, 31, 144–149. [Google Scholar] [CrossRef]
- Harita, Y.; Urae, S.; Akashio, R.; Isojima, T.; Miura, K.; Yamada, T.; Yamamoto, K.; Miyasaka, Y.; Furuyama, M.; Takemura, T.; et al. Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome. Eur. J. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Lu, X.Y.; Liu, B.C.; Cao, Y.Z.; Song, C.; Su, H.; Chen, G.; Klein, J.D.; Zhang, H.X.; Wang, L.H.; Ma, H.P. High glucose reduces expression of podocin in cultured human podocytes by stimulating TRPC6. Am. J. Physiol. Ren. Physiol. 2019, 317, F1605–F1611. [Google Scholar] [CrossRef]
- Forst, A.L.; Olteanu, V.S.; Mollet, G.; Wlodkowski, T.; Schaefer, F.; Dietrich, A.; Reiser, J.; Gudermann, T.; Mederos y Schnitzler, M.; Storch, U. Podocyte Purinergic P2X4 Channels Are Mechanotransducers That Mediate Cytoskeletal Disorganization. J. Am. Soc. Nephrol. 2016, 27, 848–862. [Google Scholar] [CrossRef]
- Anderson, M.; Kim, E.Y.; Hagmann, H.; Benzing, T.; Dryer, S.E. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol. Cell Physiol. 2013, 305, C276–C289. [Google Scholar] [CrossRef]
- Cormont, M.; Metón, I.; Mari, M.; Monzo, P.; Keslair, F.; Gaskin, C.; McGraw, T.E.; Le Marchand-Brustel, Y. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 2003, 4, 97–112. [Google Scholar] [CrossRef]
- Yaddanapudi, S.; Altintas, M.M.; Kistler, A.D.; Fernandez, I.; Möller, C.C.; Wei, C.; Peev, V.; Flesche, J.B.; Forst, A.L.; Li, J.; et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Investig. 2011, 121, 3965–3980. [Google Scholar] [CrossRef]
- Löwik, M.M.; Groenen, P.J.T.A.; Pronk, I.; Lilien, M.R.; Goldschmeding, R.; Dijkman, H.B.; Levtchenko, E.N.; Monnens, L.A.; van den Heuvel, L.P. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007, 72, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Bareke, E.; Takeda, N.; Aoudjit, L.; Baldwin, C.; Pisano, P.; Matsuda, J.; El Andalousi, J.; Muhtadie, L.; Bernard, C.; et al. Recessive mutation in CD2AP causes focal segmental glomerulosclerosis in humans and mice. Kidney Int. 2019, 95, 57–61. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, T.J.; Anthony, E.C.; Hensbergen, P.J.; Deelder, A.M.; Hordijk, P.L. Rac1 recruits the adapter protein CMS/CD2AP to cell-cell contacts. J. Biol. Chem. 2010, 285, 20137–20146. [Google Scholar] [CrossRef]
- Balbas, M.D.; Burgess, M.R.; Murali, R.; Wongvipat, J.; Skaggs, B.J.; Mundel, P.; Weins, A.; Sawyers, C.L. MAGI-2 scaffold protein is critical for kidney barrier function. Proc. Natl. Acad. Sci. USA 2014, 111, 14876–14881. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Asanuma, K.; Fukuda, T.; Ohwada, S.; Yoshida, M.; Nishimori, K. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. Am. J. Pathol. 2014, 184, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Empitu, M.A.; Kadariswantiningsih, I.N.; Aizawa, M.; Asanuma, K. MAGI-2 and scaffold proteins in glomerulopathy. Am. J. Physiol. Ren. Physiol. 2018, 315, F1336–F1344. [Google Scholar] [CrossRef]
- Ashraf, S.; Kudo, H.; Rao, J.; Kikuchi, A.; Widmeier, E.; Lawson, J.A.; Tan, W.; Hermle, T.; Warejko, J.K.; Shril, S.; et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat. Commun. 2018, 9, 1960. [Google Scholar] [CrossRef]
- Lehtonen, S.; Ryan, J.J.; Kudlicka, K.; Iino, N.; Zhou, H.; Farquhar, M.G. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc. Natl. Acad. Sci. USA 2005, 102, 9814–9819. [Google Scholar] [CrossRef]
- Kanai, M.; Jeon, H.; Ojima, M.; Nishino, T.; Usui, T.; Yadav, M.K.; Kulathunga, K.; Morito, N.; Takahashi, S.; Hamada, M. Phenotypic analysis of mice carrying human-type MAFB p.Leu239Pro mutation. Biochem. Biophys. Res. Commun. 2020, 523, 452–457. [Google Scholar] [CrossRef]
- Zankl, A.; Duncan, E.L.; Leo, P.J.; Clark, G.R.; Glazov, E.A.; Addor, M.-C.; Herlin, T.; Kim, C.A.; Leheup, B.P.; McGill, J.; et al. Multicentric Carpotarsal Osteolysis Is Caused by Mutations Clustering in the Amino-Terminal Transcriptional Activation Domain of MAFB. Am. J. Hum. Genet. 2012, 90, 494–501. [Google Scholar] [CrossRef]
- Sato, Y.; Tsukaguchi, H.; Morita, H.; Higasa, K.; Tran, M.T.N.; Hamada, M.; Usui, T.; Morito, N.; Horita, S.; Hayashi, T.; et al. A mutation in transcription factor MAFB causes Focal Segmental Glomerulosclerosis with Duane Retraction Syndrome. Kidney Int. 2018, 94, 396–407. [Google Scholar] [CrossRef]
- Srivastava, T.; Garola, R.E.; Whiting, J.M.; Alon, U.S. Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int. 2001, 59, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kistler, A.; Faridi, M.H.; Meyer, J.O.; Tryniszewska, B.; Mehta, D.; Yue, L.; Dryer, S.; Reiser, J. Synaptopodin Limits TRPC6 Podocyte Surface Expression and Attenuates Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3308–3319. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Tsuruya, K.; Yotsueda, H.; Tokumoto, M.; Ikeda, H.; Katafuchi, R.; Fujimi, S.; Hirakata, H.; Iida, M. Expression of synaptopodin and GLEPP1 as markers of steroid responsiveness in primary focal segmental glomerulosclerosis. Life Sci. 2006, 79, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wang, Z.; Pan, X.; Wang, W.; Chen, X.; Ren, H.; Hao, C.; Han, B.; Chen, N. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2010, 25, 824–835. [Google Scholar] [CrossRef]
- Tian, X.; Ishibe, S. Targeting the podocyte cytoskeleton: From pathogenesis to therapy in proteinuric kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodríguez-Pérez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Meng, L.; Cao, S.; Lin, N.; Zhao, J.; Cai, X.; Liang, Y.; Huang, K.; Lin, M.; Chen, X.; Li, D.; et al. Identification of a Novel ACTN4 Gene Mutation Which Is Resistant to Primary Nephrotic Syndrome Therapy. Biomed Res. Int. 2019, 2019, 5949485. [Google Scholar] [CrossRef]
- Henderson, J.M.; Al-Waheeb, S.; Weins, A.; Dandapani, S.V.; Pollak, M.R. Mice with altered alpha-actinin-4 expression have distinct morphologic patterns of glomerular disease. Kidney Int. 2008, 73, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Kos, C.H.; Le, T.C.; Sinha, S.; Henderson, J.M.; Kim, S.H.; Sugimoto, H.; Kalluri, R.; Gerszten, R.E.; Pollak, M.R. Mice deficient in alpha-actinin-4 have severe glomerular disease. J. Clin. Investig. 2003, 111, 1683–1690. [Google Scholar] [CrossRef]
- Gee, H.Y.; Saisawat, P.; Ashraf, S.; Hurd, T.W.; Vega-Warner, V.; Fang, H.; Beck, B.B.; Gribouval, O.; Zhou, W.; Diaz, K.A.; et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Investig. 2013, 123, 3243–3253. [Google Scholar] [CrossRef]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Gupta, I.R.; Takano, T. Loss of Rho-GDIα sensitizes podocytes to lipopolysaccharide-mediated injury. Am. J. Physiol. Ren. Physiol. 2015, 308, F1207–F1216. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Zhang, F.; Ashraf, S.; Kohl, S.; Sadowski, C.E.; Vega-Warner, V.; Zhou, W.; Lovric, S.; Fang, H.; Nettleton, M.; et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Investig. 2015, 125, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Cong, E.H.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot–Marie–Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Chun, J.; Perez-Gill, C.; Yan, P.; Stillman, I.E.; Higgs, H.N.; Alper, S.L.; Schlöndorff, J.S.; Pollak, M.R. FSGS-Causing INF2 Mutation Impairs Cleaved INF2 N-Fragment Functions in Podocytes. J. Am. Soc. Nephrol. 2020, 31, 374–391. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Tonna, S.J.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef]
- Gbadegesin, R.A.; Lavin, P.J.; Hall, G.; Bartkowiak, B.; Homstad, A.; Jiang, R.; Wu, G.; Byrd, A.; Lynn, K.; Wolfish, N.; et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. 2012, 81, 94–99. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.S.; McNagny, K.M. The role of podocalyxin in health and disease. J. Am. Soc. Nephrol. 2009, 20, 1669–1676. [Google Scholar] [CrossRef]
- Doyonnas, R.; Kershaw, D.B.; Duhme, C.; Merkens, H.; Chelliah, S.; Graf, T.; McNagny, K.M. Anuria, omphalocele, and perinatal lethality in mice lacking the CD34-related protein podocalyxin. J. Exp. Med. 2001, 194, 13–27. [Google Scholar] [CrossRef]
- Kang, H.G.; Lee, M.; Lee, K.B.; Hughes, M.; Kwon, B.S.; Lee, S.; McNagny, K.M.; Ahn, Y.H.; Ko, J.M.; Ha, I.S.; et al. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp. Mol. Med. 2017, 49, e414. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Wu, R.M.; Ho, C.H.; Cheng, J.; Lin, H.Y.; Lin, C.H. Genetic analysis of PODXL gene in patients with familial and young-onset Parkinson’s disease in a Taiwanese population. Neurobiol. Aging 2019, 84, 235.e9–235.e10. [Google Scholar] [CrossRef] [PubMed]
- El-Ashmawy, H.M.; Selim, F.O.; Hosny, T.A.M.; Almassry, H.N. Association of serum podocalyxin levels with peripheral arterial disease in patients with type 2 diabetes. J. Diabetes Complicat. 2019, 33, 495–499. [Google Scholar] [CrossRef]
- Mansilla, M.; Wang, Y.; Hyett, J.; da Silva Costa, F.; Nie, G. Serum podocalyxin for early detection of preeclampsia at 11-13 weeks of gestation. Placenta 2018, 71, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef]
- Riehle, M.; Büscher, A.K.; Gohlke, B.-O.; Kaßmann, M.; Kolatsi-Joannou, M.; Bräsen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef]
- Santín, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; García-Maset, R.; Giménez, I.; Ruíz, P.; Mendizábal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 3089–3096. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Möller, C.C.; Du, J.; Yue, L.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef]
- Zhu, B.; Chen, N.; Wang, Z.H.; Pan, X.X.; Ren, H.; Zhang, W.; Wang, W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 2009, 664, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jirka, G.; Rosenberg, P.B.; Buckley, A.F.; Gomez, J.A.; Fields, T.A.; Winn, M.P.; Spurney, R.F. Gq signaling causes glomerular injury by activating TRPC6. J. Clin. Investig. 2015, 125, 1913–1926. [Google Scholar] [CrossRef]
- Freichel, M.; Vennekens, R.; Olausson, J.; Stolz, S.; Philipp, S.E.; Weissgerber, P.; Flockerzi, V. Functional role of TRPC proteins in native systems: Implications from knockout and knock-down studies. J. Physiol. 2005, 567, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Hinkes, B.; Wiggins, R.C.; Gbadegesin, R.; Vlangos, C.N.; Seelow, D.; Nürnberg, G.; Garg, P.; Verma, R.; Chaib, H.; Hoskins, B.E.; et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006, 38, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Ashraf, S.; Tan, W.; van der Ven, A.T.; Gee, H.Y.; Braun, D.A.; Fehér, K.; George, S.P.; Esmaeilniakooshkghazi, A.; Choi, W.I.; et al. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 4257–4269. [Google Scholar] [CrossRef]
- Yu, S.; Choi, W.I.; Choi, Y.J.; Kim, H.Y.; Hildebrandt, F.; Gee, H.Y. PLCE1 regulates the migration, proliferation, and differentiation of podocytes. Exp. Mol. Med. 2020, 52, 594–603. [Google Scholar] [CrossRef]
- Atchison, D.K.; O’Connor, C.L.; Menon, R.; Otto, E.A.; Ganesh, S.K.; Wiggins, R.C.; Smrcka, A.V.; Bitzer, M. Hypertension induces glomerulosclerosis in phospholipase C-ε1 deficiency. Am. J. Physiol. Ren. Physiol. 2020, 318, F1177–F1187. [Google Scholar] [CrossRef]
- Hashmi, J.A.; Safar, R.A.; Afzal, S.; Albalawi, A.M.; Abdu-Samad, F.; Iqbal, Z.; Basit, S. Whole exome sequencing identification of a novel insertion mutation in the phospholipase C ε-1 gene in a family with steroid resistant inherited nephrotic syndrome. Mol. Med. Rep. 2018, 18, 5095–5100. [Google Scholar] [CrossRef]
- Kim, Y.H.; Goyal, M.; Wharram, B.; Wiggins, J.; Kershaw, D.; Wiggins, R. GLEPP1 receptor tyrosine phosphatase (Ptpro) in rat PAN nephrosis. A marker of acute podocyte injury. Nephron 2002, 90, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Wharram, B.L.; Goyal, M.; Gillespie, P.J.; Wiggins, J.E.; Kershaw, D.B.; Holzman, L.B.; Dysko, R.C.; Saunders, T.L.; Samuelson, L.C.; Wiggins, R.C. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Investig. 2000, 106, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Ozaltin, F.; Ibsirlioglu, T.; Taskiran, E.Z.; Baydar, D.E.; Kaymaz, F.; Buyukcelik, M.; Kilic, B.D.; Balat, A.; Iatropoulos, P.; Asan, E.; et al. Disruption of PTPRO Causes Childhood-Onset Nephrotic Syndrome. Am. J. Hum. Genet. 2011, 89, 139–147. [Google Scholar] [CrossRef]
- Kim, Y.H.; Goyal, M.; Kurnit, D.; Wharram, B.; Wiggins, J.; Holzman, L.; Kershaw, D.; Wiggins, R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001, 60, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, H.P.; Mao, Y.Y.; Jin, J.; Chen, J.H. Reduced glomerular epithelial protein 1 expression and podocyte injury in immunoglobulin A nephropathy. J. Int. Med. Res. 2007, 35, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol. 2013, 9, 200–210. [Google Scholar] [CrossRef]
- Pozzi, A.; Jarad, G.; Moeckel, G.W.; Coffa, S.; Zhang, X.; Gewin, L.; Eremina, V.; Hudson, B.G.; Borza, D.B.; Harris, R.C.; et al. Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev. Biol. 2008, 316, 288–301. [Google Scholar] [CrossRef]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef]
- Verma, R.; Venkatareddy, M.; Kalinowski, A.; Patel, S.R.; Garg, P. Integrin Ligation Results in Nephrin Tyrosine Phosphorylation In Vitro. PLoS ONE 2016, 11, e0148906. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Mitra, S.K.; Ilic, D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta (Bba) Mol. Cell Res. 2004, 1692, 77–102. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Lausecker, F.; Tian, X.; Inoue, K.; Wang, Z.; Pedigo, C.E.; Hassan, H.; Liu, C.; Zimmer, M.; Jinno, S.; Huckle, A.L.; et al. Vinculin is required to maintain glomerular barrier integrity. Kidney Int. 2018, 93, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.-A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef]
- Tian, X.; Kim, J.J.; Monkley, S.M.; Gotoh, N.; Nandez, R.; Soda, K.; Inoue, K.; Balkin, D.M.; Hassan, H.; Son, S.H.; et al. Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J. Clin. Investig. 2014, 124, 1098–1113. [Google Scholar] [CrossRef]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef]
- Ramachandran, R.; Schmid, S.L. The dynamin superfamily. Curr. Biol. 2018, 28, R411–R416. [Google Scholar] [CrossRef]
- Yamada, H.; Abe, T.; Satoh, A.; Okazaki, N.; Tago, S.; Kobayashi, K.; Yoshida, Y.; Oda, Y.; Watanabe, M.; Tomizawa, K.; et al. Stabilization of actin bundles by a dynamin 1/cortactin ring complex is necessary for growth cone filopodia. J. Neurosci. 2013, 33, 4514–4526. [Google Scholar] [CrossRef]
- Gu, C.; Yaddanapudi, S.; Weins, A.; Osborn, T.; Reiser, J.; Pollak, M.; Hartwig, J.; Sever, S. Direct dynamin-actin interactions regulate the actin cytoskeleton. Embo J. 2010, 29, 3593–3606. [Google Scholar] [CrossRef]
- Schiffer, M.; Teng, B.; Gu, C.; Shchedrina, V.A.; Kasaikina, M.; Pham, V.A.; Hanke, N.; Rong, S.; Gueler, F.; Schroder, P.; et al. Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat. Med. 2015, 21, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Schafer, D.A. Dynamin: Expanding its scope to the cytoskeleton. Int. Rev. Cell Mol. Biol. 2013, 302, 187–219. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Chang, J.; Gu, C. Dynamin rings: Not just for fission. Traffic 2013, 14, 1194–1199. [Google Scholar] [CrossRef]
- Wu, W.; Kitamura, S.; Truong, D.M.; Rieg, T.; Vallon, V.; Sakurai, H.; Bush, K.T.; Vera, D.R.; Ross, R.S.; Nigam, S.K. Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. Am. J. Physiol. Ren. Physiol. 2009, 297, F210–F217. [Google Scholar] [CrossRef] [PubMed]
- Karamatic Crew, V.; Burton, N.; Kagan, A.; Green, C.A.; Levene, C.; Flinter, F.; Brady, R.L.; Daniels, G.; Anstee, D.J. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood 2004, 104, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Donnan, M.D.; Scott, R.P.; Onay, T.; Tarjus, A.; Onay, U.V.; Quaggin, S.E. Genetic Deletion of Emp2 Does Not Cause Proteinuric Kidney Disease in Mice. Front. Med. (Lausanne) 2019, 6, 189. [Google Scholar] [CrossRef]
- Dorval, G.; Gribouval, O.; Martinez-Barquero, V.; Machuca, E.; Tête, M.J.; Baudouin, V.; Benoit, S.; Chabchoub, I.; Champion, G.; Chauveau, D.; et al. Clinical and genetic heterogeneity in familial steroid-sensitive nephrotic syndrome. Pediatr. Nephrol. 2018, 33, 473–483. [Google Scholar] [CrossRef]
- Wan, X.; Chen, Z.; Choi, W.-I.; Gee, H.Y.; Hildebrandt, F.; Zhou, W. Loss of Epithelial Membrane Protein 2 Aggravates Podocyte Injury via Upregulation of Caveolin-1. J. Am. Soc. Nephrol. Jasn 2016, 27, 1066–1075. [Google Scholar] [CrossRef]
- Hagel, M.; George, E.L.; Kim, A.; Tamimi, R.; Opitz, S.L.; Turner, C.E.; Imamoto, A.; Thomas, S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell Biol. 2002, 22, 901–915. [Google Scholar] [CrossRef]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef]
- Chinthalapudi, K.; Rangarajan, E.S.; Izard, T. The interaction of talin with the cell membrane is essential for integrin activation and focal adhesion formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10339–10344. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wu, C. ILK: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 2012, 24, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bordoy, R.; Stanchi, F.; Moser, M.; Braun, A.; Kudlacek, O.; Wewer, U.M.; Yurchenco, P.D.; Fässler, R. PINCH1 regulates cell-matrix and cell-cell adhesions, cell polarity and cell survival during the peri-implantation stage. J. Cell Sci. 2005, 118, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, C.; Ma, P.; Lai, Y.; Yang, F.; Cai, J.; Cheng, Z.; Zhang, K.; Liu, Z.; Tian, Y.; et al. Kindlin-2 Association with Rho GDP-Dissociation Inhibitor α Suppresses Rac1 Activation and Podocyte Injury. J. Am. Soc. Nephrol. 2017, 28, 3545–3562. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Iatropoulos, P.; Donadelli, R.; Calabria, A.; Maranta, R.; Cassis, P.; Buelli, S.; Tomasoni, S.; Piras, R.; Krendel, M.; et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 295–306. [Google Scholar] [CrossRef]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef]
- Tabibzadeh, N.; Fleury, D.; Labatut, D.; Bridoux, F.; Lionet, A.; Jourde-Chiche, N.; Vrtovsnik, F.; Schlegel, N.; Vanhille, P. MYH9-related disorders display heterogeneous kidney involvement and outcome. Clin. Kidney J. 2019, 12, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Lane, B.M.; Khan, K.; Pediaditakis, I.; Xiao, J.; Wu, G.; Wang, L.; Kovalik, M.E.; Chryst-Stangl, M.; Davis, E.E.; et al. The Human FSGS-Causing ANLN R431C Mutation Induces Dysregulated PI3K/AKT/mTOR/Rac1 Signaling in Podocytes. J. Am. Soc. Nephrol. 2018, 29, 2110–2122. [Google Scholar] [CrossRef] [PubMed]


