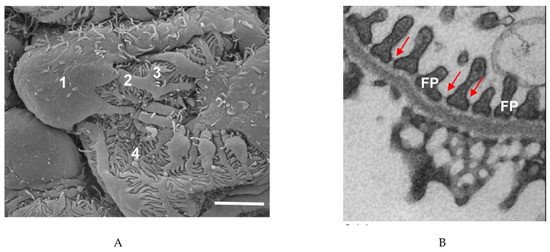
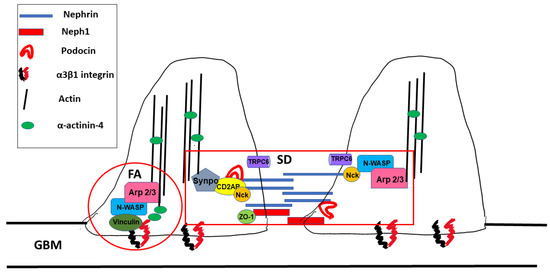
3.6.1. Mutations Affecting Nephrin and Podocin
Given the critical role that nephrin plays in maintenance of the SD, it is not surprising that mutations affecting the nephrin gene,
NPHS1, are associated with the severe kidney disease congenital nephrotic syndrome of the Finnish type [
26,
33,
35,
84,
85,
86,
87]. Additionally, mutations that alter nephrin trafficking or signaling have also been implicated as causing disease. Examples include mutations in the genes
GTPase activating protein and VPS9 domains 1 (GAPVD1) and
ankyrin repeat and FYVE domain containing 1 (ANKFY1) which were identified to cause a recessive type of nephrotic syndrome by impairing nephrin trafficking [
88].
Podocin, another crucial protein of the slit diaphragm that interacts with nephrin and CD2AP, has also been implicated in human disease. Mutations in
NPHS2, which codes for podocin, and
LIM homeobox transcription factor 1 Beta (LMX1B), a regulator of podocin expression, have been found to cause nephrotic syndrome [
89,
90,
91,
92,
93]. Since podocin expression is limited to the glomeruli,
NPHS2 mutations typically present without significant extrarenal manifestations [
92,
93,
94]. However
LMX1B mutations are causative in the syndromic disease nail–patella syndrome characterized by hypoplastic nails and patella, skeletal deformities and varying degrees of nephropathy [
89,
90,
95]. The underlying pathophysiology caused by mutations affecting podocin relates to impaired nephrin signaling due to the inability of nephrin to associate with lipid rafts as well as podocin’s effects on TRPC6 [
52,
82,
96,
97,
98].
3.6.2. Mutations Affecting Adaptor Proteins/Signal Integrators
CD2AP is scaffolding protein which links nephrin and podocin with the actin cytoskeleton [
56,
99,
100]. Mutations of CD2AP gene have resulted in nephrotic syndrome and FSGS lesions in humans [
56,
101,
102]. The mechanistic cause for disease is still being studied but is believed to be due to CD2AP’s interactions with F-actin through CAPZ as well as its interactions with Rac1 and synaptopodin [
15,
55,
100,
103]. Similar to CD2AP, membrane-associated guanylate kinase 2 (MAGI2) is another scaffolding protein that interacts with the nephrin complex and helps maintain the functional structure of the slit diaphragm [
104,
105]. Mutations in
MAGI2 have been found to cause nephrotic syndrome in mice and humans [
106]. MAGI2-associated disease is caused in part due to its interaction with tachykinin 2 (TAC2), which interacts with the Rho GTPase deleted in liver cancer 1 (DLC1), as mentioned above, as well as its interaction with α-actinin-4 and synaptopodin [
15,
107,
108].
As demonstrated in the
LMX1B mutations causing kidney disease, diseases can arise if transcriptional regulators of slit diaphragm proteins are mutated. MAF BZIP transcription factor B (MAFB) is an important example of this fact as it is a transcription factor that regulates podocin, nephrin, and CD2AP expression [
109]. Two mutations in the
MAFB gene have been identified as causative in two different syndromic diseases where proteinuria is a feature, multicentric carpotarsal osteolysis [
110] and focal segmental glomerulosclerosis with Duane retraction syndrome (FSGS-DRS) [
111].
Synaptodopodin is intriguing from a disease modulation standpoint since it regulates actin activity through its effects on α-actinin-4 and through RhoA activation and Cdc42 inhibition [
67]. Synaptopodin has been shown to have anti-proteinuric effects and its absence has been noted in several different disease states [
68,
112,
113,
114]. Furthermore, mutations affecting synaptodopodin expression have been noted in patients with proteinuric kidney disease [
115]. A significant amount of research is aimed at examining how to modulate synaptopodin for treatment benefit since synaptopodin has TRPC6 modifying properties and part of the effectiveness of the anti-proteinuric effects of cyclosporin A is due to modification of synaptopodin [
113,
116]
α-actinin-4 is critical for bundling F-actin but also links integrins that adhere to the basement membrane to the cytoskeleton [
16,
18]. Mutations affecting the
ACTN4 gene, which codes for α-actinin-4, clinically present as nephrotic syndrome with FSGS but have a wide age range in which disease manifests [
115,
117,
118]. Notably, different mutations in the
ACTN4 genes have demonstrated that the absence of α-actinin-4 as well as mutations causing increased affinity for F-actin, both lead to disease [
117,
118,
119,
120].
As outlined, regulation of the cytoskeleton dynamic is maintained by a complex balance through RhoA, Rac1, and Cdc42. Given this complexity and need for a balance between all these pathways, numerous gene mutations associated with this system have been identified as causing diseases. Mutations in
Rho GDP dissociation Inhibitor alpha (ARHGDIA)
and Rho-GTPase activating protein 24 (ARHGAP24) have both been shown to result in increased Rac1 and Cdc42 activity resulting in disease [
121,
122,
123]. Similarly, mutations in
KN motif and ankyrin repeat domains 1/2/4 (KANK1,
KANK2, and
KANK4) have all been reported to be associated with nephrotic syndrome due to their interactions with proteins that interact with RhoA (KANK 1 interacts with synaptopodin and KANK2 interacts with ARHGDIA) [
124]. Mutations in
FAT1,
intersectin 1 (
ITSN1), and
ITSN2 genes have all been reported to cause nephrotic syndrome due to their adverse effects on Cdc42 activation resulting in impaired podocyte migration [
107,
125]. One of the most studied mutations affecting Cdc42 are the mutations in
inverted formin 2 (INF2) which codes for a formin protein and has been found to cause Charcot–Marie–Tooth disease as well as FSGS without Charcot–Marie–Tooth disease [
126,
127,
128,
129,
130,
131]. Subsequently, mutations in the
INF2 gene have been found to be one of the most common causes in familial nephrotic syndrome [
130,
131].
DLC1 is another example of a Rho GTPase-activating protein in which mutations cause nephrotic syndrome [
107]. Since DLC1 is regulated by cyclic dependent kinase 20 (CDK20) and tensin-2 (TNS2), mutations in
TNS2 and
cyclin-dependent kinase 20 (CDK20) are also causative in some cases of nephrotic syndrome [
107].
Podocalyxin is a cell surface sialomucin expressed by several different cell types including podocytes, vascular endothelium, hematopoietic cells and some subsets of neurons, that is important for podocyte actin cytoskeleton remodeling [
132]. Podocalyxin’s C-terminal binding motif (DTHL) interacts with Na
+/H
+ exchanger regulator proteins (NHERF1 and NHERF2) and actin-binding protein ezrin, and can modify the cytoskeleton through these interactions [
132]. When podocalyxin is knocked down, podocytes develop abnormal cell morphology. Knockout of the
PODXL gene in mice results in loss of foot processes and loss of the slit diaphragm entirely causing a phenotype of anuric renal failure, omphalocele, and perinatal death [
133]. A reported loss-of-function mutation in
PODXL has been reported clinically with a very similar phenotype to the one observed in PODXL knockout mice [
134]. More recently, podocalyxin has been studied outside the kidney for its role in other diseases like cancer, neurologic and atherosclerotic diseases [
132,
135,
136,
137].
TPRC6 plays multiple roles in activating RhoA and Rac1 in response to mechanical stress and influences the function of nephrin, podocin, CD2AP and synaptopodin by regulating intracellular calcium [
138]. TPRC6 mutations have also been noted in several cases of congenital FSGS [
75,
139,
140,
141]. Interestingly, disease severity caused by TRPC6 mutations seems to be dependent on the degree to which they alter intracellular calcium [
138,
142]. Most of these mutations are gain-of-function mutations mediated in part through angiotensin II that causes excessive intracellular calcium [
77,
138,
139]. There have been mutations reported that result in a loss of function of TRPC6 which cause disease as well [
140]. Remarkably, disease presentation can vary widely in age of onset and severity including some families with members with proteinuric disease and some without disease, despite having the same mutation [
143]. As such, it has been postulated that a “second hit” is needed to manifest disease. This theory is supported by a transgenic mouse with a TRPC6 mutation that has no evidence of disease at baseline but developed more severe disease after injury than wild type animals [
144]. Since TRPC6 is regulated in part by angiotensin II, a significant amount of research is being done to investigate its role in modulating non-congenital glomerular diseases such as diabetic nephropathy and autoimmune glomerulonephritis [
138]. Studies looking at modulating TRPC6 in other disease states have yielded some intriguing results [
138], but additional studies are needed to determine when and how to optimally alternate TRPC6 signaling in order to derive clinical benefit since both over and under activation of the channel can be associated with disease.
Phospholipase Cε (PLCε1), a cytoplasmic phospholipase, has been found to be important in podocytes due to its function of regulating internal calcium balance by releasing of calcium from internal stores as well as through activating TRPC6 [
65,
145]. As mentioned, intracellular calcium affects nephrin and synaptopodin expression in addition to RhoA activation [
146,
147,
148]. Mutations in PLCε1 have been reported to cause SRNS because of its effects on nephrin, synaptopodin, TRPC6 and RhoA [
146,
149,
150].
Glomerular epithelial protien-1 (GLEPP1) is a tyrosine phosphatase expressed on podocyte foot processes and suggested to regulate glomerular pressure and affects nephrin content at the slit diaphragm [
151,
152]. Decreases in GLEPP-1 have been reported in several diseases including a recessive form of nephrotic kidney disease associated with a mutation on the
GLEPP-1 gene,
PTPRO [
114,
151,
153,
154,
155]. Intriguingly, PTPRO knockout mice were reported to be non-proteinuric at baseline but did have significant microscopic abnormalities in podocyte structure and spacing [
152].