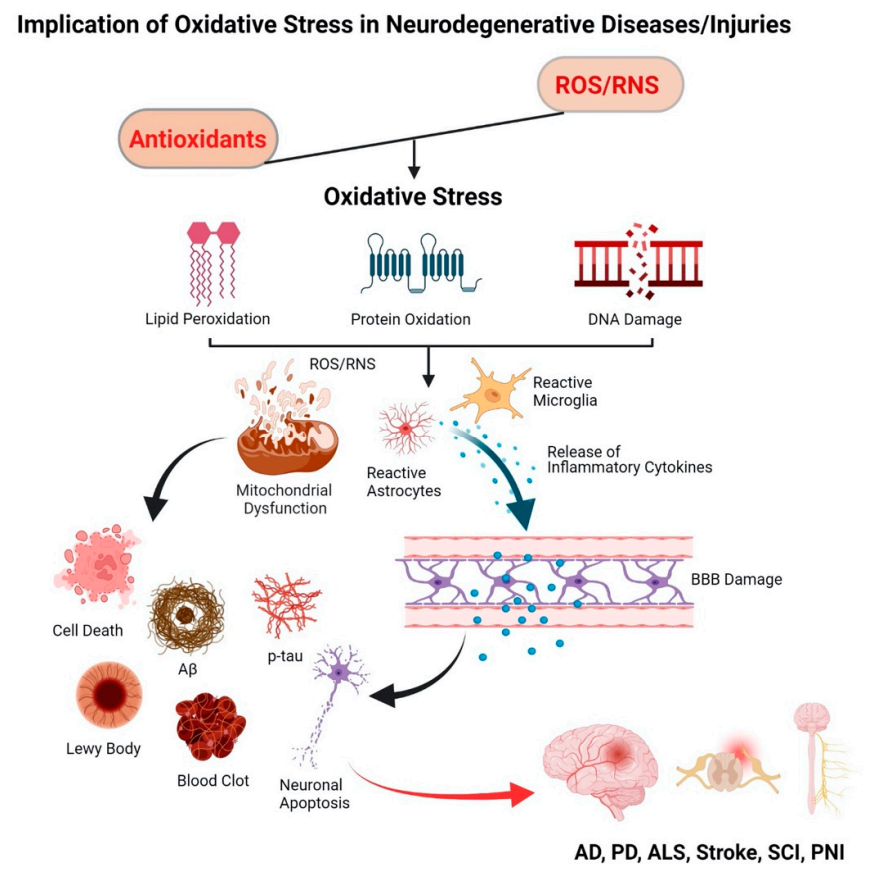
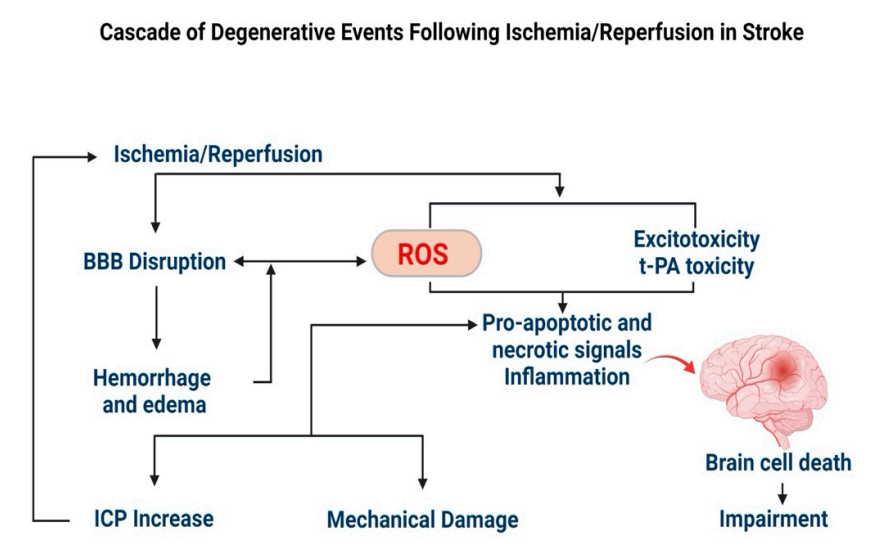
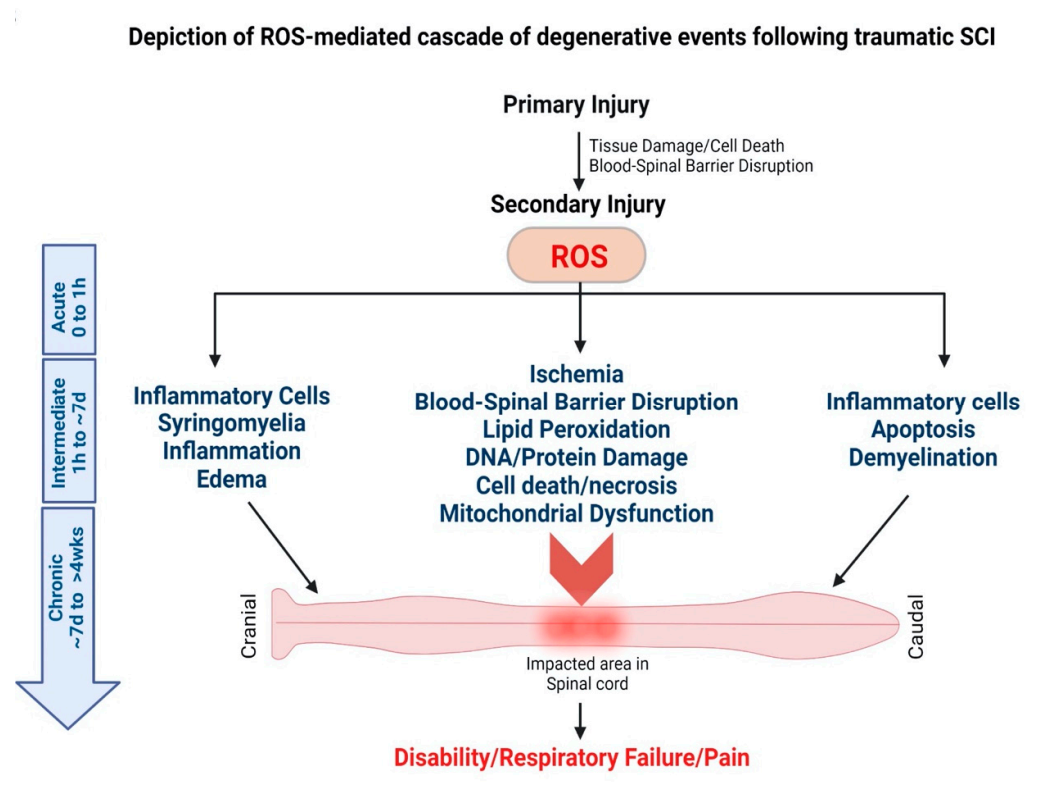
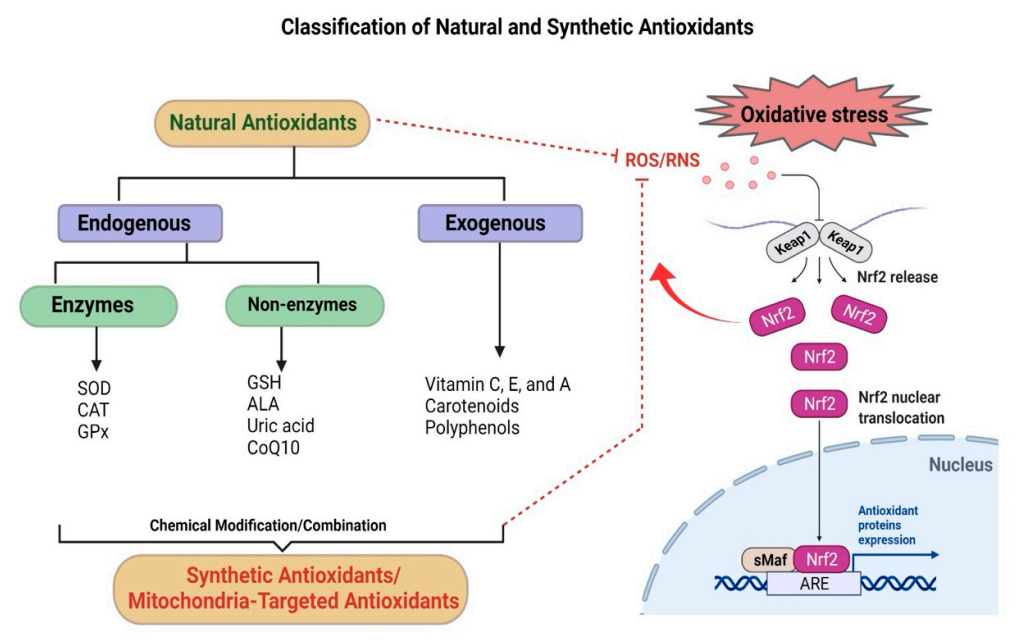
AD, a leading cause of dementia, is characterized by a progressive decline in cognitive function
[48]. Amyloid beta (Aβ) plaques, neurofibrillary tangles (NFTs), hyperphosphorylated microtubule-associated protein tau, and neuronal loss within the brain are specific histopathological hallmarks of AD patients
[49]. Prior to the development of plaque pathology, oxidative stress has been recognized as the key player in the etiology of AD, contributing to mitochondrial dysfunction in synapses and neurons, and in Aβ production
[50][51]. In fact, the concept of oxidative stress in AD was originally derived from the “free radical theory of aging”, meaning that free radicals play a central role in the aging process
[52]. Mitochondrial dysfunction in AD includes impaired mitochondrial complexes
[53][54][55][56], malfunctioning of F1Fo adenosine triphosphate (ATP) synthase, which is involved in oxidative phosphorylation
[57][58], and damage to the promoter of the mitochondrial ATP synthase gene that controls ATP generation
[59][60]. Further, dysfunctional mitochondria produce 4-HNE that upregulates γ-secretase complex and promotes cleavage of the amyloid precursor protein (APP), leading to Aβ accumulation
[61][62]. In addition, increased Ca
2+ and ROS levels lead to a buildup of p-tau aggregates which are toxic and are considered as one of the defining pathological hallmarks of AD patients
[63]. ROS also play a pivotal role in the stress kinases like the phospho-c-Jun N-terminal kinase 1 (p-JNK) pathway which is linked to tau hyperphosphorylation and cell death in response to Aβ accumulation
[64]. Further, oxidative stress reduces the activities of antioxidants, i.e., superoxide dismutase (SOD), catalase (CAT), and glutathione S-transferase (GST), thus weakening the endogenous antioxidant defense of the CNS
[65]. Further, oxidative stress increases The increased levels of LPO under oxidative stress are strongly associated with neurotoxicity in AD
[50] as it leads to an increase in amyloidogenesis through upregulation of β-secretase expression
[66]. Although there are several downstream degenerative events, it appears that mitochondrial dysfunction and oxidative stress are the key triggering factors in the pathogenesis of AD.