1000/1000
Hot
Most Recent
 +1 point
+1 point
Records of eDNA/eRNA exposome may reflect the early appearance, persistence, and presence of biotic and/or abiotic-exposure-mediated modifications in these nucleic acid molecules. Functional genome- and epigenome-wide mapping of eDNA offer great promise to help elucidate the human exposome. Assessment of longitudinal exposure to physical, biological, and chemical agents present in the environment through eDNA/eRNA may enable the building of an integrative causal dynamic stochastic model to estimate environmental causes of human health deficits. Development and validation of monitoring of eDNA/eRNA exposome should seriously be considered to introduce into safety and risk assessment and as surrogates of chronic exposure to environmental stressors.
The organismal genetic material, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA) present in the environment (home, work or recreational built environment, hazardous wastes, lake and river water, air, etc.) refer to environmental DNA or RNA (eDNA/eRNA). All organisms, including microbes, animals, and humans leave behind traces of genetic material in the environment as they pass through habitats or interact with other organisms. Besides entire microorganism genomes (bacteria, virus, fungus and protozoans), the sources of eDNA/eRNA could be from human or animal blood, hair, excrements (urine, feces, mucous, saliva, secretions), skin, sperm, eggs, roots, leaves, fruit and pollen of plants, and rotting bodies of larger organisms.The eDNA/eRNA-based exposome assessment is expected to rely on our ability to capture the genome- and epigenome-wide signatures left behind by individuals in the indoor and outdoor physical spaces through shedding, excreting, etc.
Humans are stochastically exposed from conception/origin onwards to multiple sets of environmental factors present in the indoor and outdoor physical spaces where we live, recreate, and work. Exposure to these different sets of environments creates an overlapping human exposome. To clarify the environmental exposure overlaps, the exposome is categorized into three broad environmental domains [1]. The first environmental domain consists of a common external environment which may include the rural/urban environment (e.g., buildings, furnishings, open and public spaces, roads, utilities, and other infrastructure which either introduce, allow, or restrict environmental factors) and climate factors. This domain is followed with a specific external environment exposure in the indoor and outdoor physical spaces of specific toxins, pathogens, radiation, lifestyle factors (e.g., tobacco, alcohol), diet, physical activity, etc. Finally, the third domain consists of the internal environment resulting from the overall exposure of both common and specific environments. This domain may include internal biological factors such as metabolic factors, hormones, gut microflora, inflammation, oxidative stress, etc. These comprehensive and complete descriptions of dynamic environmental domains which individuals are exposed to on a regular basis indicate the complexity of cumulative multiple exposures of environmental stressors and their interactions in real-life human environmental settings (summarized in Figure 1). Exposures to these overlapping environmental external and internal factors are largely responsible for the majority of non-hereditary sporadic chronic diseases.
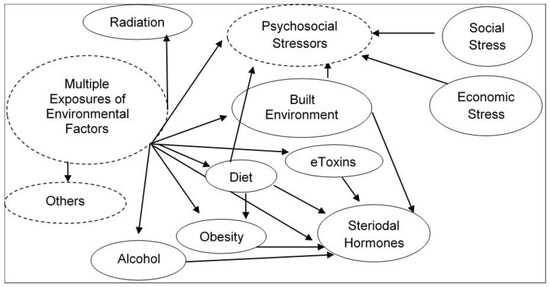
Figure 1. Illustration of complexity of cumulative multiple exposures of environmental stressors and their interactions in real-life human environmental settings.
Comprehensive analyses of entire complex and dynamic environmental exposure to multiple stressors over time require high sampling effort and multi-species approaches [2]. Existing environmental exposure monitoring approaches are inefficient, not cost effective, destructive, and taxonomically or functionally biased. Also, there is a major reliance on professional taxonomic identification or organismal sample collections and analysis that are not translatable to real environmental settings. There is no fully validated and optimized tool available for mapping an entire exposome for an individual.
Accurate assessment of the exposome through estimating several hundreds of time-varying exposures has become a major challenge. High time resolution human exposome evaluation is currently being pursued using high-throughput omics technologies to integrate a wide range of exposures. However, the major hurdle for these approaches is accurately assessing the past exposure. These genetic ‘breadcrumbs’ left behind, shed, or excreted from organisms, can serve as important biological materials for assessing the human exposome and health responses. This can be achieved by monitoring changes in the genomic and epigenomic marks in these eDNA/eRNA samples in relation to their environment [3]. The basis for this concept comes from the recent studies showing the genetic change through time recorded in the ancient DNA (aDNA), including sequence damage and reconstruction of temporal change in many large vertebrates from extreme climate cycles of paleoenvironments [4][5]. Like aDNA, the eDNA/eRNA are also to able keep records of long-lasting genetic and epigenetic changes from environmental insults and thus, they have potential to serve as a proxy of past and present exposure of multiple stressors. Previous eDNA monitoring has been mostly limited to the investigations related to studying the impact of the environment on biodiversity and ecological health [6]. Recently, there has been a rapid advancement in the application of eDNA/eRNA in biomonitoring of diseases, population genetics, and biosecurity [7]. Studies evaluating responses to environmental exposure to hazardous agents are mostly limited to monitoring areas with known pollutants or in controlled situations to determine differences in community composition. Study of how eDNA from microbes, plants, and animals that have changed and/or adapted over time can also serve as a record of exposure of environmental stressors over time. For example, ambient air, soil, rivers, and lakes, are continuously exposed to multiple stressors over time, including hazardous agent, anthropogenic activity, and changes in climate. There is an urgent need to analyze human eDNA to assess exposome effects on human health based on eDNA signatures observed in field conditions.
Hazardous outcomes caused by environmental stressors to ecological health are determined not only by toxic properties, but also changes in the ecosystems, including indigenous biodiversity and community composition. eDNA is one of the most emerging critical biological resources in the field of ecotoxicology [11]. Sequencing and bioinformatic analysis of eDNA samples have been extensively used to comprehensively investigate changes in the ecosystem and use it as a barometer of ecological health [10][12]. Therefore, here we briefly discuss it. Biodiversity monitoring is the standard for environmental impact assessment of anthropogenic activities. eDNA sequencing provides a powerful lens to detect alterations or modifications in biodiversity [13]. High-throughput sequencing (HTS) of eDNA metabarcoding of different ribosomal bacterial and eukaryotic markers is used to measure biotic index values, the taxonomic resolution of molecular markers, and measuring their performance to assess the environmental impact on biodiversity and metabolic function of numerous communities, including soils, ocean, human guts, oral cavity [14][15][16][17]. Most of the ecological and evolutionary metagenomics studies using sequencing of targeted marker genes, such as 16S/18S rDNA/rRNA, offer very limited knowledge about intraspecies diversity and no functional information.
The eDNA has also been used for the detection of rare species, indicator species, or invasive species. This is accomplished by comparing a highly variable region as a barcode in the conserved regions in DNA among species to discriminate between species and determine the taxonomy. To determine various species, metabarcoding eDNA approaches and targeted assays have also been used on ice cores, dirt from swamps, archaeological sites, seawater, fecal samples, and herbal medicines. The application of eDNA metabarcoding extends across many applications. This has provided a huge new tool and more relevant approaches for assessments of risks posed to ecosystems by hazardous agents [11]. Researchers are additionally able to sample organisms and life stages which elude traditional techniques. For example, juvenile and gamete DNA are included in the detection of eDNA from water or sediments. eDNA analysis enables monitoring of the presence of the great crested newt—a protected species in Europe—and detecting invasive bighead carp in the Great Lakes, USA [18][19].
The efforts of studying air pollution are mostly focused on particulate matters in the ambient air, whereas inhalable microorganisms in polluted air have not been well investigated, and the few reported studies mostly focused on taxonomic identification rather than pathogenic potential of microbes. Next-generation sequencing (NGS) of eDNA is not only able to detect invasive species, pathogenic microbes, and responses to pollution, but it can also assess air quality, environmental exposure to chemicals, and their implications in human health [20][21][22][23]. For example, in the case of a major environment insults, such as leaching of toxins from hazardous waste sites, oil spill or intrusion of invasive species, eDNA sequencing and their analysis have been applied to detect what species have been impacted and what genomic changes have occurred in these species over time [24][25][26]. The presence of respiratory microbial allergens and pathogens in outdoors air samples from Beijing collected during a severe smog event and in hospital air samples have been detected by metabarcoding eDNA approaches [27][28]. In summary, these studies provide support, indicating that biodiversity detected by NGS of eDNA can also assess the health risk associated from exposure to environmental agents.





