Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jinsong Zhang | + 2109 word(s) | 2109 | 2021-11-04 07:39:42 | | | |
| 2 | Vivi Li | Meta information modification | 2109 | 2021-11-16 07:49:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, J. Glutamate Protects against Catecholamine Oxidation. Encyclopedia. Available online: https://encyclopedia.pub/entry/16027 (accessed on 07 February 2026).
Zhang J. Glutamate Protects against Catecholamine Oxidation. Encyclopedia. Available at: https://encyclopedia.pub/entry/16027. Accessed February 07, 2026.
Zhang, Jinsong. "Glutamate Protects against Catecholamine Oxidation" Encyclopedia, https://encyclopedia.pub/entry/16027 (accessed February 07, 2026).
Zhang, J. (2021, November 16). Glutamate Protects against Catecholamine Oxidation. In Encyclopedia. https://encyclopedia.pub/entry/16027
Zhang, Jinsong. "Glutamate Protects against Catecholamine Oxidation." Encyclopedia. Web. 16 November, 2021.
Copy Citation
Catecholamines, such as dopamine and norepinephrine, take part in regulating a variety of mental processes, including cognitive ability, attention, memory, mood, and reward. Glutamate, as a neurotransmitter, plays an important role in learning, memory, neuronal plasticity, and brain development. The excessive stimulation of glutamate receptors causes the excitatory toxicity of neuron cells; thus, neurons are endowed with high-affinity glutamate transporters to enrich glutamate.
catecholamines
dopamine
glutamate
neurotransmitters
oxidation
1. Introduction
Catecholamines, such as dopamine and norepinephrine, take part in regulating a variety of mental processes, including cognitive ability, attention, memory, mood, and reward [1][2][3][4][5]. Catecholamines have adjacent hydroxyl groups on their benzene rings [6], thus making them susceptible to autoxidation, and produce hydrogen peroxide [7][8][9], semiquinone anion radicals [8][10], and quinones [11][12]. Quinones further initiate intramolecular cyclization to form the end products of neuromelanin polymers [13][14][15][16][17]. All these intermediates and end products may be toxic to neuron cells, and thus the autoxidation of catecholamines is considered to be an important mechanism of neuron cell loss in Parkinson’s disease [18][19][20][21]. Moreover, the disorder of copper homeostasis is involved in neurological diseases, such as Parkinson’s disease [22][23]. Redox-active copper can facilitate the oxidation of catecholamines via the formation of hydroxyl radicals [24][25]. It is known that DNA is an especially sensitive site within the cell for copper-mediated damage because copper ions have a high affinity for DNA, forming a DNA–Cu complex [26][27]. Catecholamines and DNA-localized copper can cause DNA damage via the site-specific attack of hydroxyl radicals on DNA [25][26].
Glutamate, as a neurotransmitter, plays an important role in learning, memory, neuronal plasticity, and brain development [28][29][30][31][32]. The excessive stimulation of glutamate receptors causes the excitatory toxicity of neuron cells [33][34]; thus, neurons are endowed with high-affinity glutamate transporters to enrich glutamate [35]. Consequently, extracellular glutamate concentrations, or interstitial fluid glutamate concentrations, are maintained in levels as low as 0.5–5 μM [36], and intracellular concentrations of glutamate reach as high as 6–12 mM [35]. In contrast, the intracellular concentrations of dopamine, the major catecholamine neurotransmitter in dopaminergic neurons, are only at the level of 0.05–0.1 mM [37][38][39]. The present study investigated whether glutamate has an impact on catecholamine oxidation in vitro. We found that glutamate was able to prevent the autoxidation of catecholamines and autoxidation-associated quinoprotein formation, the copper-mediated oxidation of catecholamines, catecholamine/copper-triggered DNA damage, and quinoprotein formation.
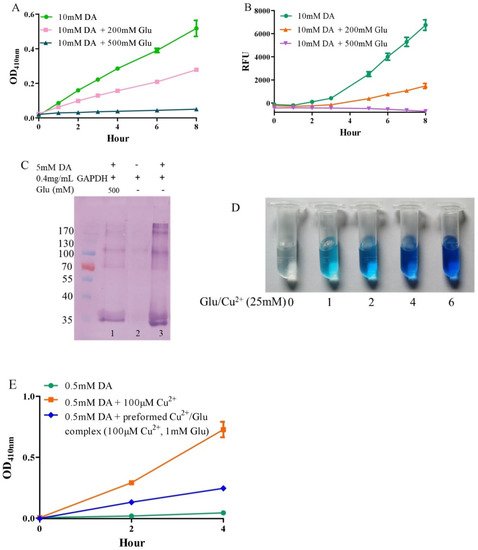
2. Autoxidation of Dopamine and the Protective Role of Glutamate
Under alkaline and aerobic conditions, dopamine can undergo autoxidation to form quinones and produce hydrogen peroxide [9][40]. The quinones further polymerize to form macromolecular neuromelanin polymers, which increase the pigmentation of neurons in the substantia nigra [41]. The pigmented dopaminergic neurons in the substantia nigra are preferentially lost in Parkinson’s disease [13]. In the present study, 10 mM of dopamine was used to observe time-dependent color development due to dopamine oxidation under physiological conditions (a pH 7.4, 0.15 M PBS). Compared to dopamine alone, the addition of −200 mM of glutamate effectively inhibited dopamine oxidation, while that of −500 mM of glutamate almost completely inhibited the oxidation, as evaluated by the color development at OD410nm (Figure 1A). Since dopamine autoxidation leads to the formation of reactive oxygen species (ROS), 50 μM of DCFH-DA as a fluorescent probe was used to detect the ROS produced by 10 mM of dopamine. The production of ROS was significantly inhibited by −200 mM of glutamate, while 500 mM of glutamate nearly entirely inhibited ROS production (Figure 1B). During the nonenzymatic autoxidation process of dopamine, intermediate quinones can covalently react with cysteine sulfhydryl groups in proteins or enzymes, leading to the formation of quinoproteins [8][42][43]. Quinoprotein adduct formation may play a role in the age-dependent selective vulnerability of dopaminergic neurons in Parkinson’s disease [44]. To detect the production of dopamine-quinoproteins, dopamine and GAPDH (with a free thiol group of a cysteine residue at position 151) were co-incubated at 37 °C for 1 h in vitro. As shown in Figure 1C, in the absence of dopamine, quinoproteins were not observed (lane 2). After a 1 h co-incubation of GAPDH and dopamine (5 mM), dopamine-quinoproteins could be clearly visualized (lane 3). The addition of glutamate (500 mM, lane 1) sufficiently inhibited quinoprotein formation. These results together suggest that glutamate has an inhibitory effect on the autoxidation of dopamine. However, the specific mechanism involved is not clear. We speculated that this may involve the nature of autoxidation. In fact, the spin limitation of dioxygen is a dynamic barrier to the oxidation of biomolecules such as dopamine [45]. The direct reaction between the two requires a large amount of activation energy; thus, the oxidation rate of biomolecules is very slow and the real autoxidation of biomolecules is a negligible process [45]. However, many transition metals with various spin states can overcome the spin limitation of dioxygen and thus increase the oxidation rate of biomolecules. The commonly described autoxidation of biomolecules such as catecholamines is actually promoted by transition metals [45]. David et al. proposed that the oxidation of dopamine in the absence of added copper may be significantly influenced by the presence of metal impurities [46]. In deionized water that was further purified by chromatography over Chelex 100 resin prior to use, epinephrine does not autoxidize. However, epinephrine was oxidized rapidly in deionized water, but this oxidation could be prevented by desferal (a potent metal chelating agent) [47]. The autoxidation of (-)-epigallocatechin-3-gallate, a well-documented redox-active catechin mainly found in green tea, can be largely prevented by EDTA, indicating that trace amounts of transition metals are involved in the autoxidation process [48]. Many buffer systems, especially phosphate, can form complexes with transition metals. Thus, in many experimental systems, the presence of trace metals in the buffer is inevitable [45]. Put simply, there is no pure “autoxidation”. The essence of biomolecule autoxidation is oxidation involving trace amounts of transition metals. It has been reported that glutamate can react with copper by forming complexes [49][50]. Indeed, as shown in Figure 1D, the characteristic blue color of glutamate-copper complexes is enhanced as a function of the increased glutamate concentrations. Preformed glutamate-copper complexes consistently show a compromised capacity to promote dopamine oxidation as compared with free copper (Figure 1E). We thus speculate that the inhibitory effect of glutamate on the autoxidation of dopamine could be attributed to glutamate’s restriction on redox-active copper bound to dopamine or the buffer system. Since trace amounts of transition metals, which promote the autoxidation of biomolecules, form complexes with biomolecules and/or buffers, as much as 14 mM of EDTA was required to effectively suppress the “autoxidation” of (-)-epigallocatechin-3-gallate [48]. The present study also consistently showed that higher molar ratios of glutamate/dopamine were needed to fully inhibit the autoxidation of dopamine. In the case of the free copper-promoted oxidation of dopamine, which has been implicated in dopamine-associated toxicity [11][51], we estimated that glutamate would be highly effective in preventing dopamine oxidation—i.e., to achieve an effective protective effect, the molar ratios of glutamate/dopamine are significantly lowered compared to the case of dopamine “autoxidation”. Next, we examined this possibility.
Figure 1. Autoxidation of dopamine and the protective role of glutamate. (A) Dopamine autoxidationmeasured by OD410 nm. (B) Dopamine-caused ROS production detected by DCFH-DA. (C) Dopamine-caused quinoprotein in GAPDH. (D) Glutamate-copper complexes. (E) Influence of free copper and preformed copper-glutamate complex on dopamine oxidation. Chemicals were mixed in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 1 h for C. Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate.
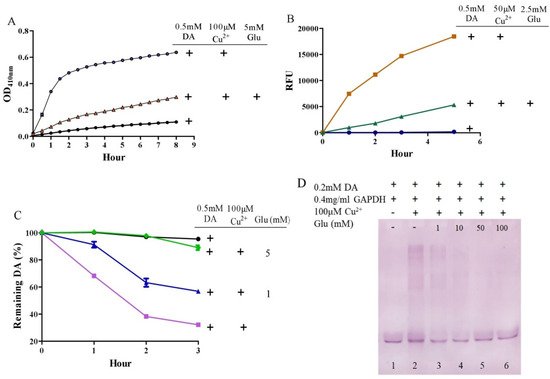
3. Glutamate Protects against Copper-Facilitated Dopamine Oxidation
It is surprising that substantial concentrations of both dopamine and copper (0.4 mM) coexist in the substantia nigra, although the precise free copper concentration is not known [46][52]. Moreover, many studies have shown that copper levels are elevated in the cerebrospinal fluid (CSF) of patients with Parkinson’s disease [53][54]. Copper can facilitate dopamine oxidation and meanwhile leads to the production of highly active hydroxyl radicals and, accordingly, DNA damage [25][55]. These processes may contribute to the observed loss of dopaminergic neurons in patients with Parkinson’s disease [56]. Therefore, we further evaluated the influence of glutamate on copper-accelerated dopamine oxidation. As shown in Figure 2A, compared with 0.5 mM of dopamine alone, the addition of 100 μM of copper hugely promoted the oxidation of dopamine. Glutamate at a concentration of 5 mM effectively suppressed copper-promoted dopamine oxidation (Figure 2A). Concerning hydroxyl radical production by copper and dopamine, we used a hydroxyl radical-specific probe, the 3-CCA, for the assessment. In a redox system of 0.5 mM of dopamine and 50 μM of copper, hydroxyl radical production was clearly sensed by the 3-CCA. In this redox system, the addition of glutamate at a concentration of only 2.5 mM substantially inhibited hydroxyl radical production (Figure 2B). In addition, we used HPLC to detect dopamine retention. As shown in Figure 2C, the copper facilitated dopamine oxidation in a time-dependent manner. After 3-h incubation of copper and 0.5 mM of dopamine, about 30% of the dopamine remained. At the same time, it was observed that the glutamate inhibited the copper-mediated dopamine oxidation in a dose-dependent manner. Specifically, the retention of dopamine could be effectively increased by as low as 1 mM of glutamate, and near-complete retention of dopamine could be achieved by 5 mM of glutamate (Figure 2C). We further characterized the influence of glutamate on dopamine-initiated quinoprotein formation (Figure 2D). Following a 20-min incubation of 0.2 mM of dopamine and GADPH, quinoproteins were hardly detected (lane 1). Under the circumstances, copper markedly promoted quinoprotein formation (lane 2). Nonetheless, glutamate was able to dose-dependently inhibit copper-initiated quinoprotein formation. A low concentration of glutamate (1 mM) could be significantly effective (lane 3), while 10 mM of glutamate almost completely inhibited copper-initiated quinoprotein formation (lane 4). Altogether, these four lines of evidence clearly demonstrate that glutamate is highly effective in inhibiting copper-facilitated dopamine oxidation.
Figure 2. Glutamate protects against copper-induced dopamine oxidation. (A) Dopamine oxidation measured by OD410 nm. (B) Hydroxyl radicals detected by 3-CCA. (C) Dopamine levels detected by HPLC. (D) Dopamine-caused quinoprotein in GAPDH. Chemicals were mixed in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 20 min for D. Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate.
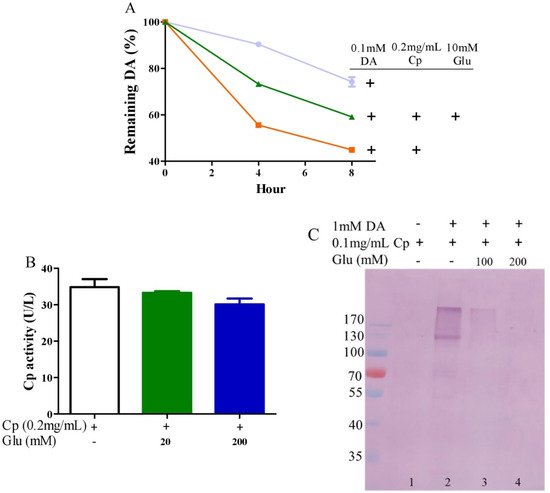
4. Glutamate Protects against Ceruloplasmin-Facilitated Dopamine Oxidation and Dopamine Oxidation-Caused Modification of Ceruloplamin
Ceruloplasmin, as expressed in human brain glial cells [57][58], is a ferrous oxidase. Ceruloplasmin plays an important role in iron homeostasis by oxidizing toxic ferrous iron so as to favor the strong binding of ferric iron to serum transferrin [59][60][61]. Ceruloplasmin with six copper atoms [62][63] also catalyzes the oxidation of catecholamines [24][47][64][65][66]. Epinephrine oxidation rates enhanced by ceruloplasmin can be slowed down by a metal chelating agent [47], suggesting that copper bound to either epinephrine or ceruloplasmin is probably involved in this catalytic reaction. We thus inferred that glutamate would be able to restrict ceruloplasmin-facilitated dopamine oxidation by forming complexes with copper. To examine this possibility, we measured dopamine oxidation catalyzed by ceruloplasmin and investigated the potential impact of glutamate on dopamine oxidation catalyzed by ceruloplasmin using HPLC. Ceruloplasmin (equivalent to 5 μM of copper) promoted the oxidation of 0.1 mM of dopamine, while 10 mM of glutamate inhibited the ceruloplasmin-catalyzed oxidation of dopamine (Figure 3A). Despite the fact that ceruloplasmin ferrous oxidase can be suppressed by a metal chelating agent such as EDTA [67], glutamate at levels that suppressed the dopamine oxidation activity of ceruloplasmin (Figure 3A) did not affect the activity of ceruloplasmin ferrous oxidase (Figure 3B). This is probably due to the different manner of copper dependence in the two types of activity. Importantly, we found that ceruloplasmin-triggered dopamine oxidation, in turn, caused the quinonization of ceruloplasmin with the formation of quinoproteins. As shown in Figure 3C, in the absence of dopamine, quinoproteins were unable to be detected from the ceruloplasmin (lane 1). In the presence of 1 mM of dopamine, the quinonization of the ceruloplasmin was salient (lane 2). Nonetheless, 100–200 mM of glutamate was highly effective in protecting against the quinonization of the ceruloplasmin (lane 3, 4). Dopamine-caused quinonization of ceruloplasmin suggests that (1) oxidized products of dopamine generated from ceruloplasmin include highly active and thus harmful quinones, and (2) the reciprocal interaction of dopamine and ceruloplasmin may impair the ferrous oxidase activity of ceruloplasmin due to quinonization, thus increasing the accumulation of ferrous ion, leading to hydroxyl radical-associated oxidative stress. Decreased ceruloplasmin levels are associated with an earlier onset of Parkinson’s disease [68][69][70]. Many studies have observed low ceruloplasmin ferrous oxidase activity in the substantia nigra and CSF of Parkinson’s disease patients [71][72][73]. However, the relevant molecular mechanism remains elusive. The interplay of dopamine and ceruloplasmin firstly identified herein may be responsible, at least in part, for the loss of the ferrous oxidase activity of ceruloplasmin. Fortunately, the reciprocal interaction of dopamine and ceruloplasmin, with a loss at both sides, can be effectively halted by glutamate.
Figure 3. Glutamate protects against ceruloplasmin-promoted dopamine oxidation. (A) Dopamine levels detected by HPLC. (B) Ferrous oxidase activity of ceruloplasmin. (C) Dopamine-caused quinoprotein in ceruloplasmin. All reactions were conducted in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 1 h for (C). Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate, Cp: Ceruloplasmin.
References
- Iuga, C.; Alvarez-Idaboy, J.R.; Vivier-Bunge, A. ROS initiated oxidation of dopamine under oxidative stress conditions in aqueous and lipidic environments. J. Phys. Chem. B 2011, 115, 12234–12246.
- Berke, J.D. What does dopamine mean? Nat. Neurosci. 2018, 21, 787–793.
- Iversen, S.D.; Iversen, L.L. Dopamine: 50 years in perspective. Trends Neurosci. 2007, 30, 188–193.
- Björklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202.
- Pignatelli, M.; Bonci, A. Role of Dopamine Neurons in Reward and Aversion: A Synaptic Plasticity Perspective. Neuron 2015, 86, 1145–1157.
- Goldstein, D.S. Catecholamines 101. Clin. Auton. Res. 2010, 20, 331–352.
- Lai, C.T.; Yu, P.H. Dopamine- and L-beta-3,4-dihydroxyphenylalanine hydrochloride (L-Dopa)-induced cytotoxicity towards catecholaminergic neuroblastoma SH-SY5Y cells. Effects of oxidative stress and antioxidative factors. Biochem. Pharmacol. 1997, 53, 363–372.
- Kalyanaraman, B.; Felix, C.C.; Sealy, R.C. Semiquinone anion radicals of catechol(amine)s, catechol estrogens, and their metal ion complexes. Environ. Health Perspect. 1985, 64, 185–198.
- Miller, J.W.; Selhub, J.; Joseph, J.A. Oxidative damage caused by free radicals produced during catecholamine autoxidation: Protective effects of O-methylation and melatonin. Free Radic. Biol. Med. 1996, 21, 241–249.
- Jodko-Piórecka, K.; Litwinienko, G. Antioxidant activity of dopamine and L-DOPA in lipid micelles and their cooperation with an analogue of α-tocopherol. Free Radic. Biol. Med. 2015, 83, 1–11.
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, Oxidative Stress and Protein-Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew. Chem. Int. Ed. Engl. 2019, 58, 6512–6527.
- Hasegawa, T. Tyrosinase-expressing neuronal cell line as in vitro model of Parkinson’s disease. Int. J. Mol. Sci. 2010, 11, 1082–1089.
- Fahn, S.; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol. 1992, 32, 804–812.
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Is Parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol. Scand. Suppl. 1989, 126, 47–54.
- Luo, Y.; Roth, G.S. The roles of dopamine oxidative stress and dopamine receptor signaling in aging and age-related neurodegeneration. Antioxid. Redox Signal. 2000, 2, 449–460.
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915.
- Banerjee, K.; Munshi, S.; Sen, O.; Pramanik, V.; Roy Mukherjee, T.; Chakrabarti, S. Dopamine Cytotoxicity Involves Both Oxidative and Nonoxidative Pathways in SH-SY5Y Cells: Potential Role of Alpha-Synuclein Overexpression and Proteasomal Inhibition in the Etiopathogenesis of Parkinson’s Disease. Parkinsons Dis. 2014, 2014, 878935.
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822.
- Li, H.-T.; Lin, D.-H.; Luo, X.-Y.; Zhang, F.; Ji, L.-N.; Du, H.-N.; Song, G.-Q.; Hu, J.; Zhou, J.-W.; Hu, H.-Y. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J. 2005, 272, 3661–3672.
- Rowe, D.B.; Le, W.; Smith, R.G.; Appel, S.H. Antibodies from patients with Parkinson’s disease react with protein modified by dopamine oxidation. J. Neurosci. Res. 1998, 53, 551–558.
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010, 30, 708–749.
- Asthana, A.; Bollapalli, M.; Tangirala, R.; Bakthisaran, R.; Mohan Rao, C. Hsp27 suppresses the Cu(2+)-induced amyloidogenicity, redox activity, and cytotoxicity of α-synuclein by metal ion stripping. Free Radic. Biol. Med. 2014, 72, 176–190.
- Bindoli, A.; Rigobello, M.P.; Deeble, D.J. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic. Biol. Med. 1992, 13, 391–405.
- Nishino, Y.; Ando, M.; Makino, R.; Ueda, K.; Okamoto, Y.; Kojima, N. Different mechanisms between copper and iron in catecholamines-mediated oxidative DNA damage and disruption of gene expression in vitro. Neurotox. Res. 2011, 20, 84–92.
- Spencer, W.A.; Jeyabalan, J.; Kichambre, S.; Gupta, R.C. Oxidatively generated DNA damage after Cu(II) catalysis of dopamine and related catecholamine neurotransmitters and neurotoxins: Role of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 139–147.
- Shao, B.; Mao, L.; Qu, N.; Wang, Y.-F.; Gao, H.-Y.; Li, F.; Qin, L.; Shao, J.; Huang, C.-H.; Xu, D.; et al. Mechanism of synergistic DNA damage induced by the hydroquinone metabolite of brominated phenolic environmental pollutants and Cu(II): Formation of DNA-Cu complex and site-specific production of hydroxyl radicals. Free Radic. Biol. Med. 2017, 104, 54–63.
- Hudspith, M.J. Glutamate: A role in normal brain function, anaesthesia, analgesia and CNS injury. Br. J. Anaesth. 1997, 78, 731–747.
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20.
- McEntee, W.J.; Crook, T.H. Glutamate: Its role in learning, memory, and the aging brain. Psychopharmacology 1993, 111, 391–401.
- Djuricić, B. Glutamate in brain: Transmitter and poison. Glas. Srp. Akad. Nauka. Med. 2002, 47, 55–76.
- Simonyi, A.; Schachtman, T.R.; Christoffersen, G.R.J. The role of metabotropic glutamate receptor 5 in learning and memory processes. Drug News Perspect. 2005, 18, 353–361.
- Egbenya, D.L.; Hussain, S.; Lai, Y.-C.; Xia, J.; Anderson, A.E.; Davanger, S. Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol. Cell. Neurosci. 2018, 92, 93–103.
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 57, 1041–1048.
- Plaitakis, A.; Shashidharan, P. Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: Implications for the pathogenesis of Parkinson’s disease. J. Neurol. 2000, 247 (Suppl. 2), II25–II35.
- Featherstone, D.E.; Shippy, S.A. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 2008, 14, 171–181.
- Takahashi, N.; Miner, L.L.; Sora, I.; Ujike, H.; Revay, R.S.; Kostic, V.; Jackson-Lewis, V.; Przedborski, S.; Uhl, G.R. VMAT2 knockout mice: Heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc. Natl. Acad. Sci. USA 1997, 94, 9938–9943.
- Yuan, J.; Callahan, B.T.; McCann, U.D.; Ricaurte, G.A. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J. Neurochem. 2001, 77, 1338–1347.
- Ponzio, F.; Achilli, G.; Calderini, G.; Ferretti, P.; Perego, C.; Toffano, G.; Algeri, S. Depletion and recovery of neuronal monoamine storage in rats of different ages treated with reserpine. Neurobiol. Aging 1984, 5, 101–104.
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467.
- Zecca, L.; Zucca, F.A.; Wilms, H.; Sulzer, D. Neuromelanin of the substantia nigra: A neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003, 26, 578–580.
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961.
- Hastings, T.G.; Zigmond, M.J. Identification of catechol-protein conjugates in neostriatal slices incubated with dopamine: Impact of ascorbic acid and glutathione. J. Neurochem. 1994, 63, 1126–1132.
- Wang, N.; Wang, Y.; Yu, G.; Yuan, C.; Ma, J. Quinoprotein adducts accumulate in the substantia nigra of aged rats and correlate with dopamine-induced toxicity in SH-SY5Y cells. Neurochem. Res. 2011, 36, 2169–2175.
- Miller, D.M.; Buettner, G.R.; Aust, S.D. Transition metals as catalysts of “autoxidation” reactions. Free Radic. Biol. Med. 1990, 8, 95–108.
- Pham, A.N.; Waite, T.D. Cu(II)-catalyzed oxidation of dopamine in aqueous solutions: Mechanism and kinetics. J. Inorg. Biochem. 2014, 137, 74–84.
- Ryan, T.P.; Miller, D.M.; Aust, S.D. The role of metals in the enzymatic and nonenzymatic oxidation of epinephrine. J. Biochem. Toxicol. 1993, 8, 33–39.
- Sang, S.; Lee, M.-J.; Hou, Z.; Ho, C.-T.; Yang, C.S. Stability of tea polyphenol (-)-epigallocatechin-3-gallate and formation of dimers and epimers under common experimental conditions. J. Agric. Food Chem. 2005, 53, 9478–9484.
- Rico-Yuson, C.A.; Hornyak, G.L.; Bora, T. Cyanide-free environment-friendly alternative to copper electroplating for zinc die-cast alloys. Environ. Sci. Pollut. Res. Int. 2021, 28, 38065–38073.
- Niu, J.; Guo, D.; Zhang, W.; Tang, J.; Tang, G.; Yang, J.; Wang, W.; Huo, H.; Jiang, N.; Cao, Y. Preparation and characterization of nanosilica copper (II) complexes of amino acids. J. Hazard. Mater. 2018, 358, 207–215.
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195.
- Stöckel, J.; Safar, J.; Wallace, A.C.; Cohen, F.E.; Prusiner, S.B. Prion protein selectively binds copper (II) ions. Biochemistry 1998, 37, 7185–7193.
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241.
- Boll, M.-C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723.
- Pattison, D.I.; Dean, R.T.; Davies, M.J. Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology 2002, 177, 23–37.
- Lévay, G.; Ye, Q.; Bodell, W.J. Formation of DNA adducts and oxidative base damage by copper mediated oxidation of dopamine and 6-hydroxydopamine. Exp. Neurol. 1997, 146, 570–574.
- Dawson, J.H.; Dooley, D.M.; Gray, H.B. Coordination environment and fluoride binding of type 2 copper in the blue copper oxidase ceruloplasmin. Proc. Natl. Acad. Sci. USA 1978, 75, 4078–4081.
- Patel, B.N.; David, S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 1997, 272, 20185–20190.
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.-P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586.
- Gutteridge, J.M. Inhibition of the Fenton reaction by the protein caeruloplasmin and other copper complexes. Assessment of ferroxidase and radical scavenging activities. Chem.-Biol. Interact. 1985, 56, 113–120.
- De Domenico, I.; Ward, D.M.; di Patti, M.C.B.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831.
- Sato, M.; Gitlin, J.D. Mechanisms of copper incorporation during the biosynthesis of human ceruloplasmin. J. Biol. Chem. 1991, 266, 5128–5134.
- Nittis, T.; Gitlin, J.D. The copper-iron connection: Hereditary aceruloplasminemia. Semin. Hematol. 2002, 39, 282–289.
- Løvstad, R.A. Activating effect of copper ions on the interaction of ceruloplasmin with catecholamines. Gen. Pharmacol. 1979, 10, 147–151.
- Rosei, M.A.; Foppoli, C.; Wang, X.T.; Coccia, R.; Mateescu, M.A. Production of melanins by ceruloplasmin. Pigment Cell Res. 1998, 11, 98–102.
- Vashchenko, G.; MacGillivray, R.T.A. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313.
- Curzon, G. The effects of some ions and chelating agents on the oxidase activity of caeruloplasmin. Biochem. J. 1960, 77, 66–73.
- Bharucha, K.J.; Friedman, J.K.; Vincent, A.S.; Ross, E.D. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson’s disease. J. Neurol. 2008, 255, 1957–1962.
- Jin, L.; Wang, J.; Zhao, L.; Jin, H.; Fei, G.; Zhang, Y.; Zeng, M.; Zhong, C. Decreased serum ceruloplasmin levels characteristically aggravate nigral iron deposition in Parkinson’s disease. Brain. 2011, 134, 50–58.
- Jin, L.; Wang, J.; Jin, H.; Fei, G.; Zhang, Y.; Chen, W.; Zhao, L.; Zhao, N.; Sun, X.; Zeng, M.; et al. Nigral iron deposition occurs across motor phenotypes of Parkinson’s disease. Eur. J. Neurol. 2012, 19, 969–976.
- Olivieri, S.; Conti, A.; Iannaccone, S.; Cannistraci, C.V.; Campanella, A.; Barbariga, M.; Codazzi, F.; Pelizzoni, I.; Magnani, G.; Pesca, M.; et al. Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J. Neurosci. 2011, 31, 18568–18577.
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.W.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 2013, 73, 554–559.
- Barbariga, M.; Curnis, F.; Andolfo, A.; Zanardi, A.; Lazzaro, M.; Conti, A.; Magnani, G.; Volontè, M.A.; Ferrari, L.; Comi, G.; et al. Ceruloplasmin functional changes in Parkinson’s disease-cerebrospinal fluid. Mol. Neurodegener. 2015, 10, 59.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
16 Nov 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No