Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Catecholamines, such as dopamine and norepinephrine, take part in regulating a variety of mental processes, including cognitive ability, attention, memory, mood, and reward. Glutamate, as a neurotransmitter, plays an important role in learning, memory, neuronal plasticity, and brain development [28,29,30,31,32]. The excessive stimulation of glutamate receptors causes the excitatory toxicity of neuron cells [33,34]; thus, neurons are endowed with high-affinity glutamate transporters to enrich glutamate [35].
- catecholamines
- dopamine
- glutamate
- neurotransmitters
- oxidation
1. Introduction
Catecholamines, such as dopamine and norepinephrine, take part in regulating a variety of mental processes, including cognitive ability, attention, memory, mood, and reward [1,2,3,4,5]. Catecholamines have adjacent hydroxyl groups on their benzene rings [6], thus making them susceptible to autoxidation, and produce hydrogen peroxide [7,8,9], semiquinone anion radicals [8,10], and quinones [11,12]. Quinones further initiate intramolecular cyclization to form the end products of neuromelanin polymers [13,14,15,16,17]. All these intermediates and end products may be toxic to neuron cells, and thus the autoxidation of catecholamines is considered to be an important mechanism of neuron cell loss in Parkinson’s disease [18,19,20,21]. Moreover, the disorder of copper homeostasis is involved in neurological diseases, such as Parkinson’s disease [22,23]. Redox-active copper can facilitate the oxidation of catecholamines via the formation of hydroxyl radicals [24,25]. It is known that DNA is an especially sensitive site within the cell for copper-mediated damage because copper ions have a high affinity for DNA, forming a DNA–Cu complex [26,27]. Catecholamines and DNA-localized copper can cause DNA damage via the site-specific attack of hydroxyl radicals on DNA [25,26].
Glutamate, as a neurotransmitter, plays an important role in learning, memory, neuronal plasticity, and brain development [28,29,30,31,32]. The excessive stimulation of glutamate receptors causes the excitatory toxicity of neuron cells [33,34]; thus, neurons are endowed with high-affinity glutamate transporters to enrich glutamate [35]. Consequently, extracellular glutamate concentrations, or interstitial fluid glutamate concentrations, are maintained in levels as low as 0.5–5 μM [36], and intracellular concentrations of glutamate reach as high as 6–12 mM [35]. In contrast, the intracellular concentrations of dopamine, the major catecholamine neurotransmitter in dopaminergic neurons, are only at the level of 0.05–0.1 mM [37,38,39]. The present study investigated whether glutamate has an impact on catecholamine oxidation in vitro. We found that glutamate was able to prevent the autoxidation of catecholamines and autoxidation-associated quinoprotein formation, the copper-mediated oxidation of catecholamines, catecholamine/copper-triggered DNA damage, and quinoprotein formation.
2. Autoxidation of Dopamine and the Protective Role of Glutamate
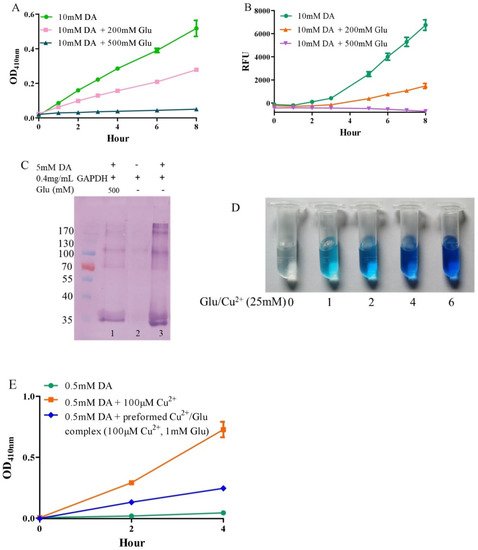
Under alkaline and aerobic conditions, dopamine can undergo autoxidation to form quinones and produce hydrogen peroxide [9,42]. The quinones further polymerize to form macromolecular neuromelanin polymers, which increase the pigmentation of neurons in the substantia nigra [43]. The pigmented dopaminergic neurons in the substantia nigra are preferentially lost in Parkinson’s disease [13]. In the present study, 10 mM of dopamine was used to observe time-dependent color development due to dopamine oxidation under physiological conditions (a pH 7.4, 0.15 M PBS). Compared to dopamine alone, the addition of −200 mM of glutamate effectively inhibited dopamine oxidation, while that of −500 mM of glutamate almost completely inhibited the oxidation, as evaluated by the color development at OD410nm (Figure 1A). Since dopamine autoxidation leads to the formation of reactive oxygen species (ROS), 50 μM of DCFH-DA as a fluorescent probe was used to detect the ROS produced by 10 mM of dopamine. The production of ROS was significantly inhibited by −200 mM of glutamate, while 500 mM of glutamate nearly entirely inhibited ROS production (Figure 1B). During the nonenzymatic autoxidation process of dopamine, intermediate quinones can covalently react with cysteine sulfhydryl groups in proteins or enzymes, leading to the formation of quinoproteins [8,44,45]. Quinoprotein adduct formation may play a role in the age-dependent selective vulnerability of dopaminergic neurons in Parkinson’s disease [46]. To detect the production of dopamine-quinoproteins, dopamine and GAPDH (with a free thiol group of a cysteine residue at position 151) were co-incubated at 37 °C for 1 h in vitro. As shown in Figure 1C, in the absence of dopamine, quinoproteins were not observed (lane 2). After a 1 h co-incubation of GAPDH and dopamine (5 mM), dopamine-quinoproteins could be clearly visualized (lane 3). The addition of glutamate (500 mM, lane 1) sufficiently inhibited quinoprotein formation. These results together suggest that glutamate has an inhibitory effect on the autoxidation of dopamine. However, the specific mechanism involved is not clear. We speculated that this may involve the nature of autoxidation. In fact, the spin limitation of dioxygen is a dynamic barrier to the oxidation of biomolecules such as dopamine [47]. The direct reaction between the two requires a large amount of activation energy; thus, the oxidation rate of biomolecules is very slow and the real autoxidation of biomolecules is a negligible process [47]. However, many transition metals with various spin states can overcome the spin limitation of dioxygen and thus increase the oxidation rate of biomolecules. The commonly described autoxidation of biomolecules such as catecholamines is actually promoted by transition metals [47]. David et al. proposed that the oxidation of dopamine in the absence of added copper may be significantly influenced by the presence of metal impurities [48]. In deionized water that was further purified by chromatography over Chelex 100 resin prior to use, epinephrine does not autoxidize. However, epinephrine was oxidized rapidly in deionized water, but this oxidation could be prevented by desferal (a potent metal chelating agent) [49]. The autoxidation of (-)-epigallocatechin-3-gallate, a well-documented redox-active catechin mainly found in green tea, can be largely prevented by EDTA, indicating that trace amounts of transition metals are involved in the autoxidation process [50]. Many buffer systems, especially phosphate, can form complexes with transition metals. Thus, in many experimental systems, the presence of trace metals in the buffer is inevitable [47]. Put simply, there is no pure “autoxidation”. The essence of biomolecule autoxidation is oxidation involving trace amounts of transition metals. It has been reported that glutamate can react with copper by forming complexes [51,52]. Indeed, as shown in Figure 1D, the characteristic blue color of glutamate-copper complexes is enhanced as a function of the increased glutamate concentrations. Preformed glutamate-copper complexes consistently show a compromised capacity to promote dopamine oxidation as compared with free copper (Figure 1E). We thus speculate that the inhibitory effect of glutamate on the autoxidation of dopamine could be attributed to glutamate’s restriction on redox-active copper bound to dopamine or the buffer system. Since trace amounts of transition metals, which promote the autoxidation of biomolecules, form complexes with biomolecules and/or buffers, as much as 14 mM of EDTA was required to effectively suppress the “autoxidation” of (-)-epigallocatechin-3-gallate [50]. The present study also consistently showed that higher molar ratios of glutamate/dopamine were needed to fully inhibit the autoxidation of dopamine. In the case of the free copper-promoted oxidation of dopamine, which has been implicated in dopamine-associated toxicity [11,53], we estimated that glutamate would be highly effective in preventing dopamine oxidation—i.e., to achieve an effective protective effect, the molar ratios of glutamate/dopamine are significantly lowered compared to the case of dopamine “autoxidation”. Next, we examined this possibility.
Figure 1. Autoxidation of dopamine and the protective role of glutamate. (A) Dopamine autoxidationmeasured by OD410 nm. (B) Dopamine-caused ROS production detected by DCFH-DA. (C) Dopamine-caused quinoprotein in GAPDH. (D) Glutamate-copper complexes. (E) Influence of free copper and preformed copper-glutamate complex on dopamine oxidation. Chemicals were mixed in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 1 h for C. Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate.
3. Glutamate Protects against Copper-Facilitated Dopamine Oxidation
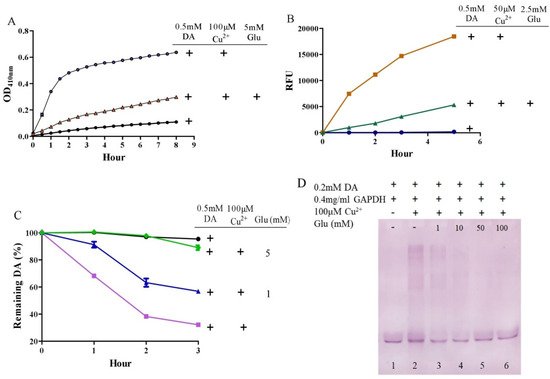
It is surprising that substantial concentrations of both dopamine and copper (0.4 mM) coexist in the substantia nigra, although the precise free copper concentration is not known [48,54]. Moreover, many studies have shown that copper levels are elevated in the cerebrospinal fluid (CSF) of patients with Parkinson’s disease [55,56]. Copper can facilitate dopamine oxidation and meanwhile leads to the production of highly active hydroxyl radicals and, accordingly, DNA damage [25,57]. These processes may contribute to the observed loss of dopaminergic neurons in patients with Parkinson’s disease [58]. Therefore, we further evaluated the influence of glutamate on copper-accelerated dopamine oxidation. As shown in Figure 2A, compared with 0.5 mM of dopamine alone, the addition of 100 μM of copper hugely promoted the oxidation of dopamine. Glutamate at a concentration of 5 mM effectively suppressed copper-promoted dopamine oxidation (Figure 2A). Concerning hydroxyl radical production by copper and dopamine, we used a hydroxyl radical-specific probe, the 3-CCA, for the assessment. In a redox system of 0.5 mM of dopamine and 50 μM of copper, hydroxyl radical production was clearly sensed by the 3-CCA. In this redox system, the addition of glutamate at a concentration of only 2.5 mM substantially inhibited hydroxyl radical production (Figure 2B). In addition, we used HPLC to detect dopamine retention. As shown in Figure 2C, the copper facilitated dopamine oxidation in a time-dependent manner. After 3-h incubation of copper and 0.5 mM of dopamine, about 30% of the dopamine remained. At the same time, it was observed that the glutamate inhibited the copper-mediated dopamine oxidation in a dose-dependent manner. Specifically, the retention of dopamine could be effectively increased by as low as 1 mM of glutamate, and near-complete retention of dopamine could be achieved by 5 mM of glutamate (Figure 2C). We further characterized the influence of glutamate on dopamine-initiated quinoprotein formation (Figure 2D). Following a 20-min incubation of 0.2 mM of dopamine and GADPH, quinoproteins were hardly detected (lane 1). Under the circumstances, copper markedly promoted quinoprotein formation (lane 2). Nonetheless, glutamate was able to dose-dependently inhibit copper-initiated quinoprotein formation. A low concentration of glutamate (1 mM) could be significantly effective (lane 3), while 10 mM of glutamate almost completely inhibited copper-initiated quinoprotein formation (lane 4). Altogether, these four lines of evidence clearly demonstrate that glutamate is highly effective in inhibiting copper-facilitated dopamine oxidation.
Figure 2. Glutamate protects against copper-induced dopamine oxidation. (A) Dopamine oxidation measured by OD410 nm. (B) Hydroxyl radicals detected by 3-CCA. (C) Dopamine levels detected by HPLC. (D) Dopamine-caused quinoprotein in GAPDH. Chemicals were mixed in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 20 min for D. Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate.
4. Glutamate Protects against Ceruloplasmin-Facilitated Dopamine Oxidation and Dopamine Oxidation-Caused Modification of Ceruloplamin
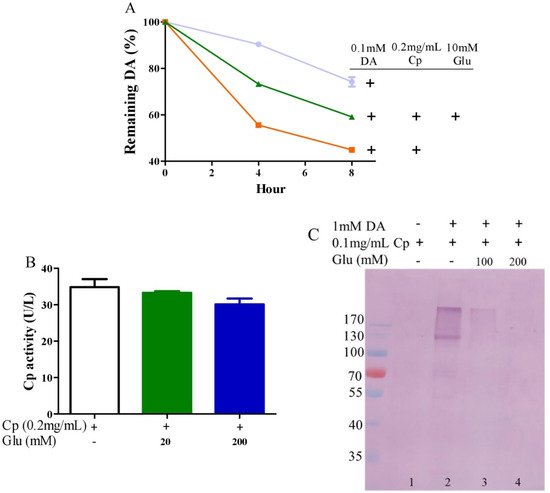
Ceruloplasmin, as expressed in human brain glial cells [59,60], is a ferrous oxidase. Ceruloplasmin plays an important role in iron homeostasis by oxidizing toxic ferrous iron so as to favor the strong binding of ferric iron to serum transferrin [61,62,63]. Ceruloplasmin with six copper atoms [64,65] also catalyzes the oxidation of catecholamines [24,49,66,67,68]. Epinephrine oxidation rates enhanced by ceruloplasmin can be slowed down by a metal chelating agent [49], suggesting that copper bound to either epinephrine or ceruloplasmin is probably involved in this catalytic reaction. We thus inferred that glutamate would be able to restrict ceruloplasmin-facilitated dopamine oxidation by forming complexes with copper. To examine this possibility, we measured dopamine oxidation catalyzed by ceruloplasmin and investigated the potential impact of glutamate on dopamine oxidation catalyzed by ceruloplasmin using HPLC. Ceruloplasmin (equivalent to 5 μM of copper) promoted the oxidation of 0.1 mM of dopamine, while 10 mM of glutamate inhibited the ceruloplasmin-catalyzed oxidation of dopamine (Figure 3A). Despite the fact that ceruloplasmin ferrous oxidase can be suppressed by a metal chelating agent such as EDTA [69], glutamate at levels that suppressed the dopamine oxidation activity of ceruloplasmin (Figure 3A) did not affect the activity of ceruloplasmin ferrous oxidase (Figure 3B). This is probably due to the different manner of copper dependence in the two types of activity. Importantly, we found that ceruloplasmin-triggered dopamine oxidation, in turn, caused the quinonization of ceruloplasmin with the formation of quinoproteins. As shown in Figure 3C, in the absence of dopamine, quinoproteins were unable to be detected from the ceruloplasmin (lane 1). In the presence of 1 mM of dopamine, the quinonization of the ceruloplasmin was salient (lane 2). Nonetheless, 100–200 mM of glutamate was highly effective in protecting against the quinonization of the ceruloplasmin (lane 3, 4). Dopamine-caused quinonization of ceruloplasmin suggests that (1) oxidized products of dopamine generated from ceruloplasmin include highly active and thus harmful quinones, and (2) the reciprocal interaction of dopamine and ceruloplasmin may impair the ferrous oxidase activity of ceruloplasmin due to quinonization, thus increasing the accumulation of ferrous ion, leading to hydroxyl radical-associated oxidative stress. Decreased ceruloplasmin levels are associated with an earlier onset of Parkinson’s disease [70,71,72]. Many studies have observed low ceruloplasmin ferrous oxidase activity in the substantia nigra and CSF of Parkinson’s disease patients [73,74,75]. However, the relevant molecular mechanism remains elusive. The interplay of dopamine and ceruloplasmin firstly identified herein may be responsible, at least in part, for the loss of the ferrous oxidase activity of ceruloplasmin. Fortunately, the reciprocal interaction of dopamine and ceruloplasmin, with a loss at both sides, can be effectively halted by glutamate.
Figure 3. Glutamate protects against ceruloplasmin-promoted dopamine oxidation. (A) Dopamine levels detected by HPLC. (B) Ferrous oxidase activity of ceruloplasmin. (C) Dopamine-caused quinoprotein in ceruloplasmin. All reactions were conducted in 0.15 M PBS (pH 7.4) and incubated at 37 °C for indicated time or 1 h for (C). Data are presented as mean ± range (n = 2). Note: DA: Dopamine, Glu: Glutamate, Cp: Ceruloplasmin.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10101564
This entry is offline, you can click here to edit this entry!