 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhongjie Fu | + 1838 word(s) | 1838 | 2021-10-26 12:03:05 | | | |
| 2 | Jessie Wu | Meta information modification | 1838 | 2021-11-09 02:34:51 | | |
Video Upload Options
Retinopathy of prematurity is defined as retinal abnormalities that occur during development as a consequence of disturbed oxygen conditions and nutrient supply after preterm birth. Both neuronal maturation and retinal vascularization are impaired, leading to the compensatory but uncontrolled retinal neovessel growth. Current therapeutic interventions target the hypoxia-induced neovessels but negatively impact retinal neurons and normal vessels. Emerging evidence suggests that metabolic disturbance is a significant and underexplored risk factor in the disease pathogenesis. Hyperglycemia and dyslipidemia correlate with the retinal neurovascular dysfunction in infants born prematurely. Nutritional and hormonal supplementation relieve metabolic stress and improve retinal maturation. Here we focus on the mechanisms through which metabolism is involved in preterm-birth-related retinal disorder from clinical and experimental investigations. We will review and discuss potential therapeutic targets through the restoration of metabolic responses to prevent disease development and progression.
1. Introduction
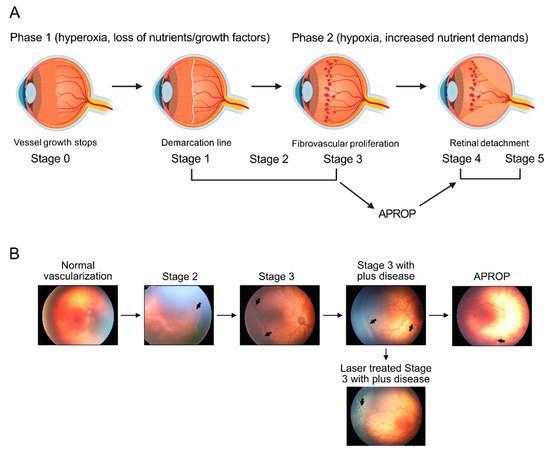
2. Regulation of Retinal Metabolism
2.1. Nutrients
2.1.1. Glucose
2.1.2. Amino Acids
2.1.3. Fatty Acids
2.2. Hormones
2.2.1. Adiponectin (APN)
2.2.2. Insulin-Growth Factor 1 (IGF-1)
References
- Hellstrom, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457.
- Fu, Z.; Gong, Y.; Lofqvist, C.; Hellstrom, A.; Smith, L.E. Review: Adiponectin in retinopathy. Biochim. Biophys. Acta 2016, 1862, 1392–1400.
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445.
- Smith, L.E.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111.
- Hansen, R.M.; Moskowitz, A.; Akula, J.D.; Fulton, A.B. The neural retina in retinopathy of prematurity. Prog. Retin. Eye Res. 2017, 56, 32–57.
- Fulton, A.B.; Dodge, J.; Hansen, R.M.; Williams, T.P. The rhodopsin content of human eyes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1878–1883.
- Akula, J.D.; Hansen, R.M.; Tzekov, R.; Favazza, T.L.; Vyhovsky, T.C.; Benador, I.Y.; Mocko, J.A.; McGee, D.; Kubota, R.; Fulton, A.B. Visual cycle modulation in neurovascular retinopathy. Exp. Eye Res. 2010, 91, 153–161.
- Lofqvist, C.A.; Najm, S.; Hellgren, G.; Engstrom, E.; Savman, K.; Nilsson, A.K.; Andersson, M.X.; Hard, A.L.; Smith, L.E.H.; Hellstrom, A. Association of Retinopathy of Prematurity With Low Levels of Arachidonic Acid: A Secondary Analysis of a Randomized Clinical Trial. JAMA Ophthalmol. 2018, 136, 271–277.
- Fu, Z.; Lofqvist, C.A.; Shao, Z.; Sun, Y.; Joyal, J.S.; Hurst, C.G.; Cui, R.Z.; Evans, L.P.; Tian, K.; SanGiovanni, J.P.; et al. Dietary omega-3 polyunsaturated fatty acids decrease retinal neovascularization by adipose-endoplasmic reticulum stress reduction to increase adiponectin. Am. J. Clin. Nutr. 2015, 101, 879–888.
- Smith, L.E. IGF-1 and retinopathy of prematurity in the preterm infant. Biol. Neonate 2005, 88, 237–244.
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58.
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244.
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203.
- Narayan, D.S.; Chidlow, G.; Wood, J.P.; Casson, R.J. Glucose metabolism in mammalian photoreceptor inner and outer segments. Clin. Exp. Ophthalmol. 2017, 45, 730–741.
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663.
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239.
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell. Metab. 2014, 19, 37–48.
- Han, X.; Kong, J.; Hartnett, M.E.; Wang, H. Enhancing Retinal Endothelial Glycolysis by Inhibiting UCP2 Promotes Physiologic Retinal Vascular Development in a Model of Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1604–1613.
- Liu, Z.; Yan, S.; Wang, J.; Xu, Y.; Wang, Y.; Zhang, S.; Xu, X.; Yang, Q.; Zeng, X.; Zhou, Y.; et al. Endothelial adenosine A2a receptor-mediated glycolysis is essential for pathological retinal angiogenesis. Nat. Commun. 2017, 8, 584.
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038.
- Fu, Z.J.; Li, S.Y.; Kociok, N.; Wong, D.; Chung, S.K.; Lo, A.C. Aldose reductase deficiency reduced vascular changes in neonatal mouse retina in oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5698–5712.
- Fu, Z.; Nian, S.; Li, S.Y.; Wong, D.; Chung, S.K.; Lo, A.C. Deficiency of aldose reductase attenuates inner retinal neuronal changes in a mouse model of retinopathy of prematurity. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 1503–1513.
- Rohlenova, K.; Goveia, J.; Garcia-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14.
- Neu, J. Glutamine supplements in premature infants: Why and how. J. Pediatr. Gastroenterol. Nutr. 2003, 37, 533–535.
- Wu, G.; Jaeger, L.A.; Bazer, F.W.; Rhoads, J.M. Arginine deficiency in preterm infants: Biochemical mechanisms and nutritional implications. J. Nutr. Biochem. 2004, 15, 442–451.
- Neu, J.; Afzal, A.; Pan, H.; Gallego, E.; Li, N.; Li Calzi, S.; Caballero, S.; Spoerri, P.E.; Shaw, L.C.; Grant, M.B. The dipeptide Arg-Gln inhibits retinal neovascularization in the mouse model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3151–3155.
- Wu, G.; Haynes, T.E.; Li, H.; Meininger, C.J. Glutamine metabolism in endothelial cells: Ornithine synthesis from glutamine via pyrroline-5-carboxylate synthase. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2000, 126, 115–123.
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352.
- Gantner, M.L.; Eade, K.; Wallace, M.; Handzlik, M.K.; Fallon, R.; Trombley, J.; Bonelli, R.; Giles, S.; Harkins-Perry, S.; Heeren, T.F.C.; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N. Engl. J. Med. 2019, 381, 1422–1433.
- Eade, K.; Gantner, M.L.; Hostyk, J.A.; Nagasaki, T.; Giles, S.; Fallon, R.; Harkins-Perry, S.; Baldini, M.; Lim, E.W.; Scheppke, L.; et al. Serine biosynthesis defect due to haploinsufficiency of PHGDH causes retinal disease. Nat. Metab. 2021, 3, 366–377.
- Vandekeere, S.; Dubois, C.; Kalucka, J.; Sullivan, M.R.; Garcia-Caballero, M.; Goveia, J.; Chen, R.; Diehl, F.F.; Bar-Lev, L.; Souffreau, J.; et al. Serine Synthesis via PHGDH Is Essential for Heme Production in Endothelial Cells. Cell Metab. 2018, 28, 573–587.e13.
- Singh, C.; Hoppe, G.; Tran, V.; McCollum, L.; Bolok, Y.; Song, W.; Sharma, A.; Brunengraber, H.; Sears, J.E. Serine and 1-carbon metabolism are required for HIF-mediated protection against retinopathy of prematurity. JCI Insight 2019, 4, e129398.
- Zhang, T.; Gillies, M.C.; Madigan, M.C.; Shen, W.; Du, J.; Grunert, U.; Zhou, F.; Yam, M.; Zhu, L. Disruption of De Novo Serine Synthesis in Muller Cells Induced Mitochondrial Dysfunction and Aggravated Oxidative Damage. Mol. Neurobiol. 2018, 55, 7025–7037.
- Zhang, T.; Zhu, L.; Madigan, M.C.; Liu, W.; Shen, W.; Cherepanoff, S.; Zhou, F.; Zeng, S.; Du, J.; Gillies, M.C. Human macular Muller cells rely more on serine biosynthesis to combat oxidative stress than those from the periphery. eLife 2019, 8, e43598.
- Becker, S.; Wang, H.; Simmons, A.B.; Suwanmanee, T.; Stoddard, G.J.; Kafri, T.; Hartnett, M.E. Targeted Knockdown of Overexpressed VEGFA or VEGF164 in Muller cells maintains retinal function by triggering different signaling mechanisms. Sci. Rep. 2018, 8, 2003.
- Le, Y.Z. VEGF production and signaling in Muller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vis. Res. 2017, 139, 108–114.
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197.
- Wei, X.; Schneider, J.G.; Shenouda, S.M.; Lee, A.; Towler, D.A.; Chakravarthy, M.V.; Vita, J.A.; Semenkovich, C.F. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 2011, 286, 2933–2945.
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009, 23, 3865–3873.
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156.
- Higuchi, A.; Ohashi, K.; Kihara, S.; Walsh, K.; Ouchi, N. Adiponectin suppresses pathological microvessel formation in retina through modulation of tumor necrosis factor-alpha expression. Circ. Res. 2009, 104, 1058–1065.
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat Commun. 2015, 6, 6228.
- Sluch, V.M.; Banks, A.; Li, H.; Crowley, M.A.; Davis, V.; Xiang, C.; Yang, J.; Demirs, J.T.; Vrouvlianis, J.; Leehy, B.; et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp(rd6) mouse. Sci. Rep. 2018, 8, 14339.
- Fu, Z.; Lofqvist, C.A.; Liegl, R.; Wang, Z.; Sun, Y.; Gong, Y.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol. Med. 2018, 10, 76–90.
- Fu, Z.; Liegl, R.; Wang, Z.; Gong, Y.; Liu, C.H.; Sun, Y.; Cakir, B.; Burnim, S.B.; Meng, S.S.; Lofqvist, C.; et al. Adiponectin Mediates Dietary Omega-3 Long-Chain Polyunsaturated Fatty Acid Protection Against Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3862–3870.
- Zhang, J.; Wang, C.; Li, L.; Man, Q.; Meng, L.; Song, P.; Froyland, L.; Du, Z.Y. Dietary inclusion of salmon, herring and pompano as oily fish reduces CVD risk markers in dyslipidaemic middle-aged and elderly Chinese women. Br. J. Nutr. 2012, 108, 1455–1465.
- Olza, J.; Mesa, M.D.; Aguilera, C.M.; Moreno-Torres, R.; Jimenez, A.; Perez de la Cruz, A.; Gil, A. Influence of an eicosapentaenoic and docosahexaenoic acid-enriched enteral nutrition formula on plasma fatty acid composition and biomarkers of insulin resistance in the elderly. Clin. Nutr. 2010, 29, 31–37.
- Kuda, O.; Jelenik, T.; Jilkova, Z.; Flachs, P.; Rossmeisl, M.; Hensler, M.; Kazdova, L.; Ogston, N.; Baranowski, M.; Gorski, J.; et al. n-3 fatty acids and rosiglitazone improve insulin sensitivity through additive stimulatory effects on muscle glycogen synthesis in mice fed a high-fat diet. Diabetologia 2009, 52, 941–951.
- Prostek, A.; Gajewska, M.; Kamola, D.; Balasinska, B. The influence of EPA and DHA on markers of inflammation in 3T3-L1 cells at different stages of cellular maturation. Lipids Health Dis. 2014, 13, 3.
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797.
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 2015, 26, 22–29.
- Kharitonenkov, A.; Larsen, P. FGF21 reloaded: Challenges of a rapidly growing field. Trends Endocrinol. Metab. 2011, 22, 81–86.
- Lin, Z.; Gong, Q.; Wu, C.; Yu, J.; Lu, T.; Pan, X.; Lin, S.; Li, X. Dynamic change of serum FGF21 levels in response to glucose challenge in human. J. Clin. Endocrinol. Metab. 2012, 97, E1224–E1228.
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63, 4057–4063.
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37.
- Fu, Z.; Gong, Y.; Liegl, R.; Wang, Z.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Saba, N.J.; Fredrick, T.W.; Morss, P.C.; et al. FGF21 Administration Suppresses Retinal and Choroidal Neovascularization in Mice. Cell Rep. 2017, 18, 1606–1613.
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Fibroblast Growth Factor 21 Protects Photoreceptor Function in Type 1 Diabetic Mice. Diabetes 2018, 67, 974–985.
- Fu, Z.; Qiu, C.; Cagnone, G.; Tomita, Y.; Huang, S.; Cakir, B.; Kotoda, Y.; Allen, W.; Bull, E.; Akula, J.D.; et al. Retinal glial remodeling by FGF21 preserves retinal function during photoreceptor degeneration. iScience 2021, 24, 102376.
- Sanchez-Infantes, D.; Gallego-Escuredo, J.M.; Diaz, M.; Aragones, G.; Sebastiani, G.; Lopez-Bermejo, A.; de Zegher, F.; Domingo, P.; Villarroya, F.; Ibanez, L. Circulating FGF19 and FGF21 surge in early infancy from infra- to supra-adult concentrations. Int. J. Obes. 2015, 39, 742–746.
- Guasti, L.; Silvennoinen, S.; Bulstrode, N.W.; Ferretti, P.; Sankilampi, U.; Dunkel, L. Elevated FGF21 leads to attenuated postnatal linear growth in preterm infants through GH resistance in chondrocytes. J. Clin. Endocrinol. Metab. 2014, 99, E2198–E2206.
- Mericq, V.; De Luca, F.; Hernandez, M.I.; Pena, V.; Rossel, K.; Garcia, M.; Avila, A.; Cavada, G.; Iniguez, G. Serum fibroblast growth factor 21 levels are inversely associated with growth rates in infancy. Horm. Res. Paediatr. 2014, 82, 324–331.
- Daughaday, W.H.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91.
- Liegl, R.; Lofqvist, C.; Hellstrom, A.; Smith, L.E. IGF-1 in retinopathy of prematurity, a CNS neurovascular disease. Early Hum. Dev. 2016, 102, 13–19.
- Cakir, B.; Hellstrom, W.; Tomita, Y.; Fu, Z.; Liegl, R.; Winberg, A.; Hansen-Pupp, I.; Ley, D.; Hellstrom, A.; Lofqvist, C.; et al. IGF1, serum glucose, and retinopathy of prematurity in extremely preterm infants. JCI Insight 2020, 5, e140363.
- Hellstrom, A.; Perruzzi, C.; Ju, M.; Engstrom, E.; Hard, A.L.; Liu, J.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Sjodell, L.; et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: Direct correlation with clinical retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2001, 98, 5804–5808.
- Hard, A.L.; Smith, L.E.; Hellstrom, A. Nutrition, insulin-like growth factor-1 and retinopathy of prematurity. Semin. Fetal Neonatal. Med. 2013, 18, 136–142.
- Hellstrom, A.; Engstrom, E.; Hard, A.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Lofqvist, C.; Svensson, E.; Holm, S.; Ewald, U.; et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics 2003, 112, 1016–1020.
- Jensen, A.K.; Ying, G.S.; Huang, J.; Quinn, G.E.; Binenbaum, G. Postnatal Serum Insulin-Like Growth Factor I and Retinopathy of Prematurity. Retina 2017, 37, 867–872.
- Hellgren, G.; Lundgren, P.; Pivodic, A.; Lofqvist, C.; Nilsson, A.K.; Ley, D.; Savman, K.; Smith, L.E.; Hellstrom, A. Decreased Platelet Counts and Serum Levels of VEGF-A, PDGF-BB, and BDNF in Extremely Preterm Infants Developing Severe ROP. Neonatology 2021, 118, 18–27.
- Smith, L.E.; Shen, W.; Perruzzi, C.; Soker, S.; Kinose, F.; Xu, X.; Robinson, G.; Driver, S.; Bischoff, J.; Zhang, B.; et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 1999, 5, 1390–1395.
- Vanhaesebrouck, S.; Daniels, H.; Moons, L.; Vanhole, C.; Carmeliet, P.; De Zegher, F. Oxygen-induced retinopathy in mice: Amplification by neonatal IGF-I deficit and attenuation by IGF-I administration. Pediatr. Res. 2009, 65, 307–310.
- Ley, D.; Hallberg, B.; Hansen-Pupp, I.; Dani, C.; Ramenghi, L.A.; Marlow, N.; Beardsall, K.; Bhatti, F.; Dunger, D.; Higginson, J.D.; et al. rhIGF-1/rhIGFBP-3 in Preterm Infants: A Phase 2 Randomized Controlled Trial. J. Pediatr. 2019, 206, 56–65.e8.
- Cakir, B.; Liegl, R.; Hellgren, G.; Lundgren, P.; Sun, Y.; Klevebro, S.; Lofqvist, C.; Mannheimer, C.; Cho, S.; Poblete, A.; et al. Thrombocytopenia is associated with severe retinopathy of prematurity. JCI Insight 2018, 3, e99448.
- Jensen, A.K.; Ying, G.S.; Huang, J.; Quinn, G.E.; Binenbaum, G. Longitudinal study of the association between thrombocytopenia and retinopathy of prematurity. J. AAPOS Off. Publ. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2018, 22, 119–123.


