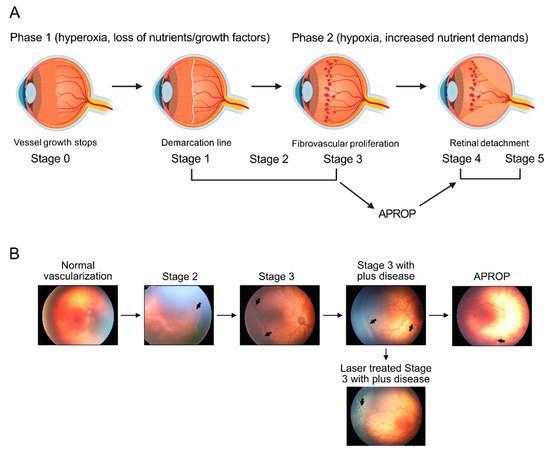
Retinopathy of prematurity is defined as retinal abnormalities that occur during development as a consequence of disturbed oxygen conditions and nutrient supply after preterm birth. Both neuronal maturation and retinal vascularization are impaired, leading to the compensatory but uncontrolled retinal neovessel growth. Current therapeutic interventions target the hypoxia-induced neovessels but negatively impact retinal neurons and normal vessels. Emerging evidence suggests that metabolic disturbance is a significant and underexplored risk factor in the disease pathogenesis. Hyperglycemia and dyslipidemia correlate with the retinal neurovascular dysfunction in infants born prematurely. Nutritional and hormonal supplementation relieve metabolic stress and improve retinal maturation. Here we focus on the mechanisms through which metabolism is involved in preterm-birth-related retinal disorder from clinical and experimental investigations. We will review and discuss potential therapeutic targets through the restoration of metabolic responses to prevent disease development and progression.
- retinal metabolism
- retinopathy of prematurity
- photoreceptor
- retinal vessel
- retinal neuron
- premature infants
- neovascularization
- oxygen-induced retinopathy
- hyperglycemia-associated retinopathy
- hyperglycemia
1. Introduction