 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marisa Granato | + 2641 word(s) | 2641 | 2021-10-25 08:27:56 | | | |
| 2 | Jason Zhu | + 693 word(s) | 3334 | 2021-10-26 04:08:28 | | |
Video Upload Options
Epstein–Barr Virus (EBV) and Kaposi’s sarcoma associated-herpesvirus (KSHV) are γ-herpesviruses that belong to the Herpesviridae family. In the last decade, many studies conducted by scientists and clinicians have indicated that nanotechnology and nanomedicine could improve the outcome of several treatments in γ-herpesvirus-associated diseases.
1. γ-Herpesviruses
1.1. Epstein–Barr Virus (EBV)
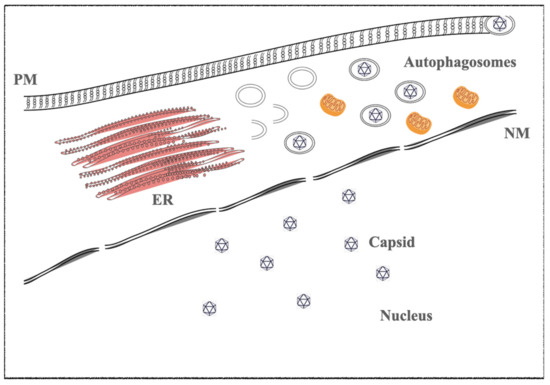
1.2. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)
2. Therapeutics Treatments in EBV- and -KSHV-Infected Malignancies
In Burkitt’s lymphoma (BL), several anti-viral agents target many proteins expressed during the late or lytic phases. Valproic acid (VPA) or valpromide (VMP), an amide derived of acid valproic, has been demonstrated to prevent the expression of immediate early EBV genes, BZLF1 and BRLF1 lytic genes. These viral proteins are necessary and essential to switch the latent to lytic phase, promoting the transcription of all genes expressed during the productive phase.
Maribavir (MBV), is approved as a therapeutic to treat human cytomegalovirus (HCMV) infection in allogeneic stem cell and bone marrow transplant recipients, is interesting, because it is also a potent inhibitor of EBV replication [26]. MBV mainly inhibits the enzymatic activity of EBV-encoded protein kinase (EBV-PK), blocking the viral DNA replication, and suppressing EBV lytic gene expression.
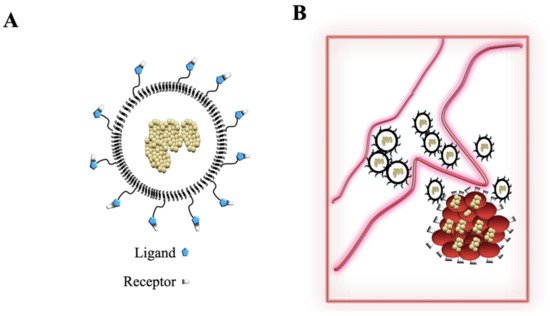
3. Nanosystems: From Liposomes to Nanoparticles (NPs)
3.1. Polymeric Nanoparticles
3.1.1. Liposomes
3.1.2. Inorganic Nanoparticles
3.1.3. Gold Nanoparticles (GNPs)
3.1.4. Silver Nanoparticles (nAg)
3.2. Nanoparticles (NPs) and EBV and KSHV Vaccines
3.3. Nanoparticles and Gamma-Herpesviruses Therapeutics
References
- Roizmann, B.; Desrosiers, R.C.; Fleckenstein, B.; Lopez, C.; Minson, A.C.; Studdert, M.J. The family Herpesviridae: An update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch. Virol. 1992, 123, 425–449.
- Turk, S.M.; Jiang, R.; Chesnokova, L.S.; Hutt-Fletcher, L.M. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 2006, 80, 9628–9633.
- Henderson, E.; Miller, G.; Robinson, J.; Heston, L. Efficiency of transformation of lymphocytes by Epstein-Barr virus. Virology 1977, 76, 152–163.
- Miller, G.; Shope, T.; Coope, D.; Waters, L.; Pagano, J.; Bornkamn, G.; Henle, W. Lymphoma in cotton-top marmosets after inoculation with Epstein-Barr virus: Tumor incidence, histologic spectrum antibody responses, demonstration of viral DNA, and characterization of viruses. J. Exp. Med. 1977, 145, 948–967.
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924.
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802.
- Morris, M.A.; Dawson, C.W.; Laverick, L.; Davis, A.M.; Dudman, J.P.; Raveenthiraraj, S.; Ahmad, Z.; Yap, L.F.; Young, L.S. The Epstein-Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFbeta and beta1 integrin signalling. Sci. Rep. 2016, 6, 19533.
- Petersson, F. Nasopharyngeal carcinoma: A review. Semin. Diagn. Pathol. 2015, 32, 54–73.
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 151–209.
- Hawkins, J.B.; Delgado-Eckert, E.; Thorley-Lawson, D.A.; Shapiro, M. The cycle of EBV infection explains persistence, the sizes of the infected cell populations and which come under CTL regulation. PLoS Pathog. 2013, 9, e1003685.
- Joseph, A.M.; Babcock, G.J.; Thorley-Lawson, D.A. EBV persistence involves strict selection of latently infected B cells. J. Immunol. 2000, 165, 2975–2981.
- Pegtel, D.M.; Subramanian, A.; Sheen, T.S.; Tsai, C.H.; Golub, T.R.; Thorley-Lawson, D.A. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: Possible role in nasopharyngeal carcinoma metastasis. J. Virol. 2005, 79, 15430–15442.
- Hurley, E.A.; Agger, S.; McNeil, J.A.; Lawrence, J.B.; Calendar, A.; Lenoir, G.; Thorley-Lawson, D.A. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J. Virol. 1991, 65, 1245–1254.
- Hurley, E.A.; Klaman, L.D.; Agger, S.; Lawrence, J.B.; Thorley-Lawson, D.A. The prototypical Epstein-Barr virus-transformed lymphoblastoid cell line IB4 is an unusual variant containing integrated but no episomal viral DNA. J. Virol. 1991, 65, 3958–3963.
- Laichalk, L.L.; Hochberg, D.; Babcock, G.J.; Freeman, R.B.; Thorley-Lawson, D.A. The dispersal of mucosal memory B cells: Evidence from persistent EBV infection. Immunity 2002, 16, 745–754.
- Tsurumi, T. EBV replication enzymes. Curr. Top. Microbiol. Immunol. 2001, 258, 65–87.
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15.
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153.
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol 2018, 1045, 377–394.
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583.
- Frappier, L. Ebna1. Curr. Top Microbiol. Immunol. 2015, 391, 3–34.
- Soni, V.; Cahir-McFarland, E.; Kieff, E. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 2007, 597, 173–187.
- Nkosi, D.; Sun, L.; Duke, L.C.; Meckes, D.G., Jr. Epstein-Barr virus LMP1 manipulates the content and functions of extracellular vesicles to enhance metastatic potential of recipient cells. PLoS Pathog. 2020, 16, e1009023.
- Nkosi, D.; Sun, L.; Duke, L.C.; Patel, N.; Surapaneni, S.K.; Singh, M.; Meckes, D.G., Jr. Epstein-Barr Virus LMP1 Promotes Syntenin-1- and Hrs-Induced Extracellular Vesicle Formation for Its Own Secretion To Increase Cell Proliferation and Migration. mBio 2020, 11, e00589-20.
- Tiwawech, D.; Srivatanakul, P.; Karalak, A.; Ishida, T. Association between EBNA2 and LMP1 subtypes of Epstein-Barr virus and nasopharyngeal carcinoma in Thais. J. Clin. Virol. 2008, 42, 1–6.
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153.
- Weiss, L.M.; Chen, Y.Y. EBER in situ hybridization for Epstein-Barr virus. Methods Mol. Biol. 2013, 999, 223–230.
- Clarke, P.A.; Sharp, N.A.; Clemens, M.J. Expression of genes for the Epstein-Barr virus small RNAs EBER-1 and EBER-2 in Daudi Burkitt’s lymphoma cells: Effects of interferon treatment. J. Gen. Virol. 1992, 73 Pt 12, 3169–3175.
- Gulley, M.L.; Glaser, S.L.; Craig, F.E.; Borowitz, M.; Mann, R.B.; Shema, S.J.; Ambinder, R.F. Guidelines for interpreting EBER in situ hybridization and LMP1 immunohistochemical tests for detecting Epstein-Barr virus in Hodgkin lymphoma. Am. J. Clin. Pathol. 2002, 117, 259–267.
- Simon, K.C.; Yang, X.; Munger, K.L.; Ascherio, A. EBNA1 and LMP1 variants in multiple sclerosis cases and controls. Acta Neurol. Scand. 2011, 124, 53–58.
- Gonnella, R.; Farina, A.; Santarelli, R.; Raffa, S.; Feederle, R.; Bei, R.; Granato, M.; Modesti, A.; Frati, L.; Delecluse, H.J.; et al. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: Interactions with BFRF1 and with the nuclear lamina. J. Virol. 2005, 79, 3713–3727.
- Lee, C.P.; Huang, Y.H.; Lin, S.F.; Chang, Y.; Chang, Y.H.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926.
- Wang, J.T.; Doong, S.L.; Teng, S.C.; Lee, C.P.; Tsai, C.H.; Chen, M.R. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 2009, 83, 1856–1869.
- Granato, M.; Farina, A.; Gonnella, R.; Santarelli, R.; Frati, L.; Faggioni, A.; Angeloni, A. Regulation of the expression of the Epstein-Barr virus early gene BFRF1. Virology 2006, 347, 109–116.
- Granato, M.; Feederle, R.; Farina, A.; Gonnella, R.; Santarelli, R.; Hub, B.; Faggioni, A.; Delecluse, H.J. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 2008, 82, 4042–4051.
- Dai, Y.C.; Liao, Y.T.; Juan, Y.T.; Cheng, Y.Y.; Su, M.T.; Su, Y.Z.; Liu, H.C.; Tsai, C.H.; Lee, C.P.; Chen, M.R. The Novel Nuclear Targeting and BFRF1-Interacting Domains of BFLF2 Are Essential for Efficient Epstein-Barr Virus Virion Release. J. Virol. 2020, 94, e01498-19.
- Feederle, R.; Neuhierl, B.; Baldwin, G.; Bannert, H.; Hub, B.; Mautner, J.; Behrends, U.; Delecluse, H.J. Epstein-Barr virus BNRF1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary B lymphocytes. J. Virol. 2006, 80, 9435–9443.
- Countryman, J.K.; Heston, L.; Gradoville, L.; Himmelfarb, H.; Serdy, S.; Miller, G. Activation of the Epstein-Barr virus BMRF1 and BZLF1 promoters by ZEBRA in Saccharomyces cerevisiae. J. Virol. 1994, 68, 7628–7633.
- Taylor, N.; Flemington, E.; Kolman, J.L.; Baumann, R.P.; Speck, S.H.; Miller, G. ZEBRA and a Fos-GCN4 chimeric protein differ in their DNA-binding specificities for sites in the Epstein-Barr virus BZLF1 promoter. J. Virol. 1991, 65, 4033–4041.
- Gradoville, L.; Grogan, E.; Taylor, N.; Miller, G. Differences in the extent of activation of Epstein-Barr virus replicative gene expression among four nonproducer cell lines stably transformed by oriP/BZLF1 plasmids. Virology 1990, 178, 345–354.
- Taylor, N.; Countryman, J.; Rooney, C.; Katz, D.; Miller, G. Expression of the BZLF1 latency-disrupting gene differs in standard and defective Epstein-Barr viruses. J. Virol. 1989, 63, 1721–1728.
- Countryman, J.; Jenson, H.; Seibl, R.; Wolf, H.; Miller, G. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J. Virol. 1987, 61, 3672–3679.
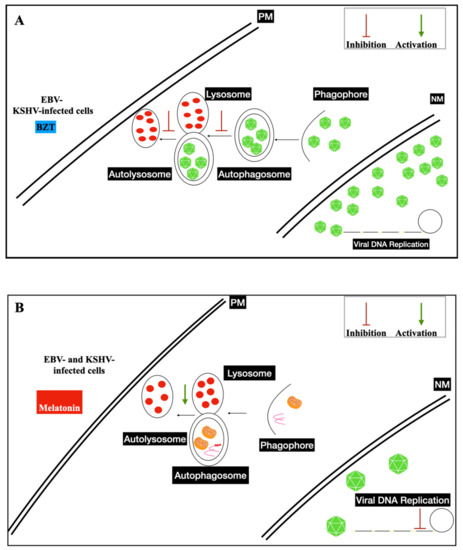
- Xia, J.; He, Y.; Meng, B.; Chen, S.; Zhang, J.; Wu, X.; Zhu, Y.; Shen, Y.; Feng, X.; Guan, Y.; et al. NEK2 induces autophagy-mediated bortezomib resistance by stabilizing Beclin-1 in multiple myeloma. Mol. Oncol. 2020, 14, 763–778.
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052.
- Granato, M.; Santarelli, R.; Filardi, M.; Gonnella, R.; Farina, A.; Torrisi, M.R.; Faggioni, A.; Cirone, M. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy 2015, 11, 1978–1986.
- Purushothaman, P.; Uppal, T.; Sarkar, R.; Verma, S.C. KSHV-Mediated Angiogenesis in Tumor Progression. Viruses 2016, 8, 198.
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV Genome Replication and Maintenance. Front. Microbiol. 2016, 7, 54.
- Ueda, K. KSHV Genome Replication and Maintenance in Latency. Adv. Exp. Med. Biol. 2018, 1045, 299–320.
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV LANA—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998.
- Wei, F.; Gan, J.; Wang, C.; Zhu, C.; Cai, Q. Cell Cycle Regulatory Functions of the KSHV Oncoprotein LANA. Front. Microbiol. 2016, 7, 334.
- Fajgenbaum, D.C. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood 2018, 132, 2323–2330.
- Gruffaz, M.; Vasan, K.; Tan, B.; Ramos da Silva, S.; Gao, S.J. TLR4-Mediated Inflammation Promotes KSHV-Induced Cellular Transformation and Tumorigenesis by Activating the STAT3 Pathway. Cancer Res. 2017, 77, 7094–7108.
- Sakakibara, S.; Tosato, G. Contribution of viral mimics of cellular genes to KSHV infection and disease. Viruses 2014, 6, 3472–3486.
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, 951.
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 2018, 17, 953–962.


