Epstein–Barr Virus (EBV) and Kaposi’s sarcoma associated-herpesvirus (KSHV) are γ-herpesviruses that belong to the Herpesviridae family. In the last decade, many studies conducted by scientists and clinicians have indicated that nanotechnology and nanomedicine could improve the outcome of several treatments in γ-herpesvirus-associated diseases.
- Epstein–Barr
- EBV
- Kaposi’s sarcoma associated-herpesvirus (KSHV)
- nanoparticles (NPs)
- γ-herpesviruses
In vivo, the primary infection is due in oropharyngeal epithelium in a productive phase, the so-called lytic infection [1,2]. The viral spread is so high and the virus infects the circulating B cells, the viral reservoir, persisting in a quiescent state (latent phase) without a release of infectious particles [3,4,5,6,7]. The biological cycle is similar to other herpesviruses. It is known that this virus infects B-lymphocytes and epithelial cells latently. During this state, the genomic DNA is tethered in a circular episome and only a set of viral proteins are expressed [8,9,10]. Many studies have been identified them as proteins necessary to establish persistent infections in target cells by reducing and inhibiting immune responses in infected individuals. Epstein–Barr nuclear antigens 1 and 2 (EBNA-1 and EBNA-2) are required to replicate the viral genome during the cell cycle progression, using the host DNA polymerase, and to gain a ‘persisting’ state, respectively [11,12,13]. In the last 60 years, several findings have showed that marmoset EBV-infected cells can proliferate and grow in vitro, expressing all of the latent proteins: six nuclear antigens (EBNA-1 to EBNA-6), latent membrane proteins (LMPs), and two non-polyadenylated RNAs (EBERs) [14,15,16,17,18,19,20]. One of them, LMP1, is an integral membrane protein that it mimics the CD40 receptor. The receptor is constitutively activated and this state induces a constitutive proliferation in infected cells. The latent proteins regulate cell cycle progression and apoptosis to avoid all of the pathways related to cellular death mechanisms.
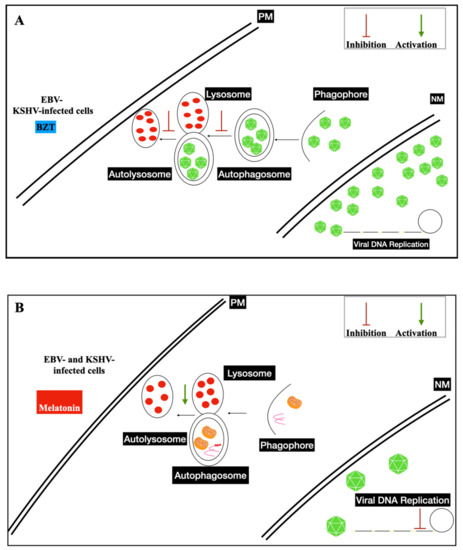
In vitro, infected cells switch from latent to lytic phases by trigging the viral replication through several drugs, such as phorbol ester (e.g., TPA-phorbol 12-myristate 13-acetate) and histone deacetylase inhibitors (e.g., sodium butyrate-NaB). The immediate early genes trigger the lytic gene activation cascade, expressing all lytic proteins necessary to structure the virions. BZT, a proteasome inhibitor, also activates the viral replication by exploiting autophagy machinery [21,22,23] ( Figure 1 ).
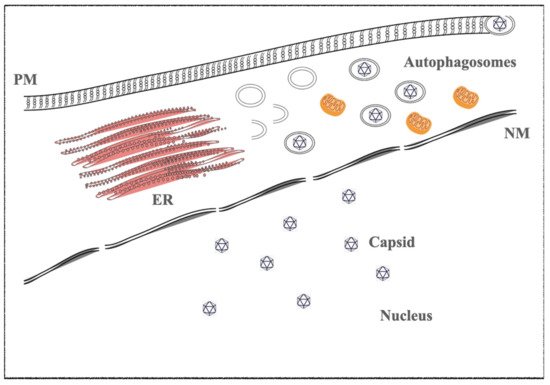
The KSHV lytic state is induced by RTA and K-bZIP proteins; they are the viral homologues of the EBV BRLF1 and BZLF1 immediate and early genes, respectively. As described in EBV, the early and late proteins are necessary to the viral replication and virion maturations [24,25,26,27].
2. Therapeutics Treatments in EBV- and -KSHV-Infected Malignancies
In Burkitt’s lymphoma (BL), several anti-viral agents target many proteins expressed during the late or lytic phases. Valproic acid (VPA) or valpromide (VMP), an amide derived of acid valproic, has been demonstrated to prevent the expression of immediate early EBV genes, BZLF1 and BRLF1 lytic genes. These viral proteins are necessary and essential to switch the latent to lytic phase, promoting the transcription of all genes expressed during the productive phase.
Maribavir (MBV), is approved as a therapeutic to treat human cytomegalovirus (HCMV) infection in allogeneic stem cell and bone marrow transplant recipients, is interesting, because it is also a potent inhibitor of EBV replication [28]. MBV mainly inhibits the enzymatic activity of EBV-encoded protein kinase (EBV-PK), blocking the viral DNA replication, and suppressing EBV lytic gene expression.
Several clinical attempts were established using the mammalian target of rapamycin inhibitor (mTOR), the key regulator of proliferation and autophagy mechanisms. Sirolimus inhibited cell growth and induced apoptosis in PEL cells, in vitro, and in a mouse xenograft model.
Increased levels of oxygen reactive species (ROS) and antioxidants have been detected in several cancer developments and progressions. Melatonin levels are altered in cancer patients due to a dysfunction in its release. This therapeutic displays anti-tumoral properties, regulating cell cycle progression, apoptosis, induced-oxidative stress, immune stimulation, and growth signaling, exerting anti-proliferative effects [29,30]. It was shown that melatonin represses the Warburg effect, ameliorates disturbed mitochondrial bioenergetics, and is a pro-oxidant in cancer cells, even in cancer stem cells (CSCs) [31,32].
3. Nanosystems: From Liposomes to Nanoparticles (NPs)
3.1. Polymeric Nanoparticles
3.1.1. Liposomes
3.1.2. Inorganic Nanoparticles
3.1.3. Gold Nanoparticles (GNPs)
3.1.4. Silver Nanoparticles (nAg)
3.2. Nanoparticles (NPs) and EBV and KSHV Vaccines
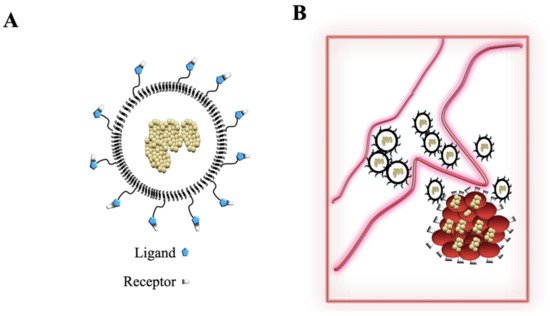
3.3. Nanoparticles and Gamma-Herpesviruses Therapeutics
REFERENCES
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48]
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111407
References
- David A. Thorley-Lawson; EBV Persistence—Introducing the Virus. Current Topics in Microbiology and Immunology 2015, 390, 151-209, 10.1007/978-3-319-22822-8_8.
- Jared B. Hawkins; Edgar Delgado-Eckert; David A. Thorley-Lawson; Michael Shapiro; The Cycle of EBV Infection Explains Persistence, the Sizes of the Infected Cell Populations and Which Come under CTL Regulation. PLoS Pathogens 2013, 9, e1003685, 10.1371/journal.ppat.1003685.
- Joseph, A.M.; Babcock, G.J.; Thorley-Lawson, D.A. EBV persistence involves strict selection of latently infected B cells. J. Immunol.
- Pegtel, D.M.; Subramanian, A.; Sheen, T.S.; Tsai, C.H.; Golub, T.R.; Thorley-Lawson, D.A. Epstein-Barr-virus-encoded LMP2A inducesinducesprimary epithelial cell migration and invasion: Possible role in nasopharyngeal carcinoma metastasis. J. Virol. 2005, 79, 15430–15442.
- Hurley, E.A.; Agger, S.; McNeil, J.A.; Lawrence, J.B.; Calendar, A.; Lenoir, G.; Thorley-Lawson, D.A. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J. Virol. 1991, 65, 1245–1254.
- Hurley, E.A.; Klaman, L.D.; Agger, S.; Lawrence, J.B.; Thorley-Lawson, D.A. The prototypical Epstein-Barr virus-transformed lympho-
- Laichalk, L.L.; Hochberg, D.; Babcock, G.J.; Freeman, R.B.; Thorley-Lawson, D.A. The dispersal of mucosal memory B cells: Evidence from persistent EBV infection. Immunity 2002, 16, 745–754.
- Tsurumi, T. EBV replication enzymes. Curr. Top. Microbiol. Immunol. 2001, 258, 65–87.
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15.
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153.
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol 2018, 1045, 377–394.
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583.
- Frappier, L. Ebna1. Curr. Top Microbiol. Immunol. 2015, 391, 3–34.
- Soni, V.; Cahir-McFarland, E.; Kieff, E. LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 2007, 597,173–187.
- Nkosi, D.; Sun, L.; Duke, L.C.; Meckes, D.G., Jr. Epstein-Barr virus LMP1 manipulates the content and functions of extracellular vesicles to enhance metastatic potential of recipient cells. PLoS Pathog. 2020, 16, e1009023.
- Nkosi, D.; Sun, L.; Duke, L.C.; Patel, N.; Surapaneni, S.K.; Singh, M.; Meckes, D.G., Jr. Epstein-Barr Virus LMP1 Promotes Syntenin-1 and Hrs-Induced Extracellular Vesicle Formation for Its Own Secretion To Increase Cell Proliferation and Migration. mBio 2020, 11,e00589-20.
- Tiwawech, D.; Srivatanakul, P.; Karalak, A.; Ishida, T. Association between EBNA2 and LMP1 subtypes of Epstein-Barr virus and nasopharyngeal carcinoma in Thais. J. Clin. Virol. 2008, 42, 1–6.
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153.
- Weiss, L.M.; Chen, Y.Y. EBER in situ hybridization for Epstein-Barr virus. Methods Mol. Biol. 2013, 999, 223–230.
- Clarke, P.A.; Sharp, N.A.; Clemens, M.J. Expression of genes for the Epstein-Barr virus small RNAs EBER-1 and EBER-2 in Daudi Burkitt’s lymphoma cells: Effects of interferon treatment. J. Gen. Virol. 1992, 73 Pt 12, 3169–3175.
- Xia, J.; He, Y.; Meng, B.; Chen, S.; Zhang, J.; Wu, X.; Zhu, Y.; Shen, Y.; Feng, X.; Guan, Y.; et al. NEK2 induces autophagy-mediated bortezomib resistance by stabilizing Beclin-1 in multiple myeloma. Mol. Oncol. 2020, 14, 763–778.
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib pro- motes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052.
- Granato, M.; Santarelli, R.; Filardi, M.; Gonnella, R.; Farina, A.; Torrisi, M.R.; Faggioni, A.; Cirone, M. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy 2015, 11, 1978–1986.
- Ueda, K. KSHV Genome Replication and Maintenance in Latency. Adv. Exp. Med. Biol. 2018, 1045, 299–320.
- Uppal, T.; Banerjee, S.; Sun, Z.; Verma, S.C.; Robertson, E.S. KSHV LANA—The master regulator of KSHV latency. Viruses 2014, 6, 4961–4998.
- Wei, F.; Gan, J.; Wang, C.; Zhu, C.; Cai, Q. Cell Cycle Regulatory Functions of the KSHV Oncoprotein LANA. Front. Microbiol. 2016, 7, 334.
- Fajgenbaum, D.C. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood 2018, 132, 2323–2330.
- Crombie, J.L.; LaCasce, A.S. Epstein Barr Virus Associated B-Cell Lymphomas and Iatrogenic Lymphoproliferative Disorders. Front. Oncol. 2019, 9, 109.
- Mehrzadi, S.; Pourhanifeh, M.H.; Mirzaei, A.; Moradian, F.; Hosseinzadeh, A. An updated review of mechanistic potentials of melatonin against cancer: Pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int. 2021, 21, 188.
- Gurunathan, S.; Qasim, M.; Kang, M.H.; Kim, J.H. Role and Therapeutic Potential of Melatonin in Various Type of Cancers. Onco Targets Ther. 2021, 14, 2019–2052.
- Jaworek, A.K.; Szepietowski, J.C.; Halubiec, P.; Wojas-Pelc, A.; Jaworek, J. Melatonin as an Antioxidant and Immunomodulator in Atopic Dermatitis-A New Look on an Old Story: A Review. Antioxidants 2021, 10, 1179.
- Guerra, J.; Devesa, J. Melatonin Exerts Anti-Inflammatory, Antioxidant, and Neuromodulatory Effects That Could Potentially Be Useful in the Treatment of Vertigo. Int. J. Otolaryngol. 2021, 2021, 6641055.
- Prieto-Dominguez, N.; Mendez-Blanco, C.; Carbajo-Pescador, S.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin enhances sorafenib actions in human hepatocarcinoma cells by inhibiting mTORC1/p70S6K/HIF-1alpha and hypoxia-mediated mitophagy. Oncotarget 2017, 8, 91402–91414.
- Shahid, S.; Prockop, S.E. Epstein-Barr virus-associated post-transplant lymphoproliferative disorders: Beyond chemotherapy treatment. Cancer Drug Resist. 2021, 4, 646–664.
- Hwang, J.; Suh, C.H.; Won Kim, K.; Kim, H.S.; Armand, P.; Huang, R.Y.; Guenette, J.P. The Incidence of Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 1785.
- van Zyl, D.G.; Mautner, J.; Delecluse, H.J. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104.
- Munz, C. Immune Control and Vaccination against the Epstein-Barr Virus in Humanized Mice. Vaccines 2019, 7, 217.
- Perez, E.M.; Foley, J.; Tison, T.; Silva, R.; Ogembo, J.G. Novel Epstein-Barr virus-like particles incorporating gH/gL-EBNA1 or gB-LMP2 induce high neutralizing antibody titers and EBV-specific T-cell responses in immunized mice. Oncotarget 2017, 8, 19255–19273.
- Ruhl, J.; Leung, C.S.; Munz, C. Vaccination against the Epstein-Barr virus. Cell Mol. Life Sci. 2020, 77, 4315–4324.
- Sun, C.; Chen, X.C.; Kang, Y.F.; Zeng, M.S. The Status and Prospects of Epstein-Barr Virus Prophylactic Vaccine Development. Front.Immunol. 2021, 12, 677027.
- Mulama, D.H.; Mutsvunguma, L.Z.; Totonchy, J.; Ye, P.; Foley, J.; Escalante, G.M.; Rodriguez, E.; Nabiee, R.; Muniraju, M.; Wussow, F.; et al. A multivalent Kaposi sarcoma-associated herpesvirus-like particle vaccine capable of eliciting high titers of neutralizing antibodies in immunized rabbits. Vaccine 2019, 37, 4184–4194.
- Kalarikkal, S.P.; Prasad, D.; Kasiappan, R.; Chaudhari, S.R.; Sundaram, G.M. A cost-effective polyethylene glycol-based method for the isolation of functional edible nanoparticles from ginger rhizomes. Sci. Rep. 2020, 10, 4456.
- Ohta, S.; Kikuchi, E.; Ishijima, A.; Azuma, T.; Sakuma, I.; Ito, T. Investigating the optimum size of nanoparticles for their delivery into the brain assisted by focused ultrasound-induced blood-brain barrier opening. Sci. Rep. 2020, 10, 18220.
- Gonzalez-Carter, D.; Liu, X.; Tockary, T.A.; Dirisala, A.; Toh, K.; Anraku, Y.; Kataoka, K. Targeting nanoparticles to the brain by exploiting the blood-brain barrier impermeability to selectively label the brain endothelium. Proc. Natl. Acad. Sci. USA 2020, 117, 19141–19150.
- Chan, H.; Kral, P. Nanoparticles Self-Assembly within Lipid Bilayers. ACS Omega 2018, 3, 10631–10637.
- You, R.; Ho, Y.S.; Hung, C.H.; Liu, Y.; Huang, C.X.; Chan, H.N.; Ho, S.L.; Lui, S.Y.; Li, H.W.; Chang, R.C. Silica nanoparticles induce neurodegeneration-like changes in behavior, neuropathology, and affect synapse through MAPK activation. Part. Fibre Toxicol. 2018, 15, 28.
- Singh, L.; Kruger, H.G.; Maguire, G.E.M.; Govender, T.; Parboosing, R. The role of nanotechnology in the treatment of viral infections. Ther. Adv. Infect. Dis. 2017, 4, 105–131. Int. J. Mol. Sci. 2021, 22, x FOR PEER REVIEW 18 of 18
- Dong, J.; Shang, Y.; Inthavong, K.; Chan, H.K.; Tu, J. Partitioning of dispersed nanoparticles in a realistic nasal passage for targeted drug delivery. Int. J. Pharm. 2018, 543, 83–95.
- Dong, J.; Shang, Y.; Inthavong, K.; Chan, H.K.; Tu, J. Partitioning of dispersed nanoparticles in a realistic nasal passage for targeted drug delivery. Int. J. Pharm. 2018, 543, 83–95.