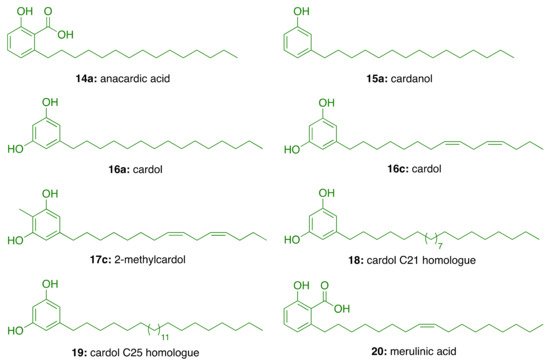
2.3.1. CNSL-Derived Rivastigmine Analogues
For this endeavor, we were inspired by the structures of rivastigmine (
1) and the alkaloid (−)-3-O-acetylspectaline (
21), obtained in large quantities from flowers and green fruits of Senna spectabilis. Given the structural similarities between
21 and saturated cardanol (
15a), this latter has been selected as a suitable starting point to develop CNSL-derived rivastigmine analogues
[33][34]. Paula et al.
[33][34] designed new potential AChE inhibitors by means of theoretical calculations. Particularly, the target compounds were modeled following a molecular hybridization strategy between
1 and cardanols (
15a). To explore structure-activity relationships, the phenolic hydroxyl of
15a was replaced by methoxy (
22), acetyl (
23), and N,N-dimethylcarbamoyl groups (
24). As for secondary amines, alicyclic and heterocyclic amines (N,N-dimethylamine (
a), N,N-diethylamine (
b), pyrrolidine (
c), and piperidine (
d)) were selected to study the conformational flexibility of this subunit. The aromatic N-methylbenzylamine (
e) was also considered for evaluating whether an additional Π-Π interaction with the Trp84 or Phe330 residues could be established (
Figure 2). The calculations of the electronic structures of the fifteen designed derivatives were initially performed at RHF level using 6-31G, 6-31G(d), 6-31+G(d) and 6-311G(d,p) base functions, then B3LYP level with the 6-31G, 6-31G (d) and 6-311+G(2d, p). Among the proposed compounds, the structures featuring the N,N-dimethycarbamoyl, N,N-dimethylamine and pyrrolidine groups showed a better correlation with
1. Based on the descriptors E (HOMO-1), E (LUMO+1), C–O56, C–NR2, E(LUMO), and ΔL+1, obtained in the principal component analysis (PCA), the N,N-dimethylcarbamates
24a–e emerged to be the most promising anticholinesterase candidates. To validate the theoretical study, the compounds were synthesized as racemic mixtures and evaluated against EeAChE. Compounds
24a–c inhibited AChE in a concentration-dependent manner, whereas
24d and
24e showed negligible activity, by inhibiting the enzyme by less than 25% at 100 μM. As predicted by theoretical studies, the N,N-dimethylamino derivative
24a turned out to be the most potent inhibitor (IC50 = 50 μM) of the series, followed by the pyrrolidinyl derivative
24c (IC50 = 84 μM), and the diethylamino
24b (IC50 = 251 μM)
[27].
Figure 2. Design of cardanol-derived AChE inhibitors by hybridization between rivastigmine (1) and (−)-3-O-acetylspectaline (21).
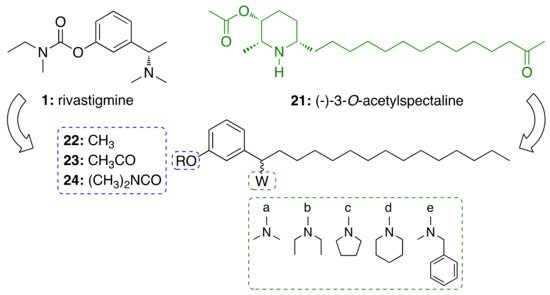
2.3.2. CNSL-Derived Dual Binding AChE Inhibitors
Motivated by the above results, we considered cardanols
15b–d as suitable frameworks for the design of AChE inhibitors with higher potency. To this end, we envisaged that combining two different pharmacophoric units could be a valuable strategy to increase activity. Particularly, we sought to link the cardanol skeleton, which might interact with the PAS through its aromatic end, with a fragment able to fish the CAS, to obtain dual binding AChE inhibitors (
Figure 3)
[35]. As a CAS key molecular feature, we selected fragments bearing a cationic head, which included a protonatable amino moiety belonging to different systems: heterocyclic amines such as pyrrolidine (
25), piperidine (
26), morpholine (
27), thiomorpholine (
28), N-substituted piperazines (
29–33), hydroxylated pyrrolidine (
34) and piperidines (
35–
37) and their corresponding O-acetyl (
38–41) and O-dimethylcarbamates (
42–
45). Since the N-ethyl-N-(2-methoxybenzyl)amino moiety has been successfully exploited by us and others to obtain powerful dual binding AChEIs
[36][37][38][39][40][41], we also synthesized hybrids
46–
48 (
Figure 3).
Figure 3. Cardanol-derived AChE inhibitors 25–48.
Collectively, twenty-four compounds were synthesized from the mixture of cardanols 15b–d and evaluated in terms of EeAChE inhibition profile at 100 µM. Of these, nineteen compounds inhibited the enzyme by more than 50%.
We report in
Table 3 the best performing derivatives, with IC50 values below 20 µM. Additionally, most of the derivatives did not show appreciable toxicity against HT-29 cells, up to a concentration of 100 μM, which indicates a potential drug-like behaviour
[35].
Table 3. Selectivity and cholinesterase inhibition of selected cardanol-derived compounds.
| Compound |
EeAChE
IC50(μM) a |
eqBChE
IC50 (μM) a |
SI b |
EeAChE
Ki (μM) c |
| 25 |
LDT148 |
19.6 |
16.4 |
0.8 |
22.4 (3) |
| 40 |
LDT149 |
16.1 |
19.0 |
1.2 |
19.0 (3) |
| 42 |
LDT154 |
13.7 |
22.8 |
1.7 |
8.6 (2) |
| 44 |
LDT150 |
14.3 |
23.5 |
1.7 |
22.4 (3) |
| 46 |
LDT167 |
17.2 |
3.1 |
0.2 |
17.0 (3) |
| 47 |
LDT161 |
6.6 |
5.0 |
0.8 |
24.4 (3) |
| 48 |
LDT160 |
16.1 |
8.0 |
0.5 |
10.4 (2) |
a Data are geometric means from 2–3 independent experiments, each performed in triplicate.
b Selectivity index: eqBChE/EeAChE IC50 ratio.
c Data are from a global fit to averaged data from 2–3 independent experiments, each performed in triplicate. Data taken from reference
[35].
Notably, compound
47, bearing a N-ethyl-N-(2-methoxybenzyl)amine moiety, showed the highest inhibitory activity against EeAChE, with a promising IC50 of 6.6 μM, and a similar inhibition profile against the human isoform (IC50 = 5.7 μM). Furthermore,
47 was predicted to cross the blood-brain barrier (BBB) by a PAMPA-BBB assay
[35]. All in all, these data suggested that the approach of obtaining potential anti-AD compounds from CNSL was worth of further pursuit and development.
To understand the different AChE inhibitory profiles of benzylamines
46–
48, Silva et al.
[42] carried out theoretical studies to identify key physicochemical properties underlying the experimental data. Particularly, electronic calculations of molecular descriptors, which included molecular volume, polarizability, polar surface area, dissociation constant (pKa), and distribution coefficient (LogD), and PCA were performed together with molecular dynamics and molecular docking. It was possible to verify that a modified cardanol (obtained from the replacement of the hydroxyl by a methoxyl group) and the protonated benzylamine derivatives assume characteristics of electron donor and electron acceptor, respectively, as revealed by electronic structure calculations. In fact, the energies of the HOMO boundary orbitals, HOMO-1, LUMO and LUMO+1, are important descriptors for measuring molecular similarities and grouping inhibitors according to their inhibitory profiles, as shown in the PCA results. Interestingly, molecular docking calculations confirmed a dual binding mode within the human AChE cavity, a characteristic that seems crucial for the stability of this type of complex
[42]. In particular, it was noted that ligands
47 and
48 are more likely to bind in an inward orientation, mimicking the donepezil X-ray pose
[42].
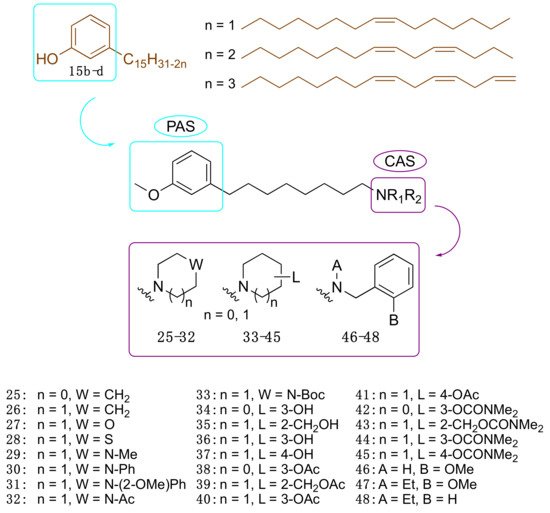
2.3.3. Cardanol-Derived Cholinesterase Inhibitors with Antioxidant and Anti-Amyloid Properties
As a further step, we were interested in the design of multifunctional CNSL-derived compounds, endowed with additional beneficial properties beyond AChE inhibition. Thus, we rationally manipulated the structure of dual binding AChE inhibitor methoxy–cardanol
47 (LDT161) (
Figure 4)
[43]. In this novel series, we kept the free phenolic group as in cardanols
15, with the objective of inhibiting Aβ aggregation and preserving the antioxidant properties, commonly found in phenols. Furthermore, we maintained a protonable aminomethyl group, critical for the inhibitory profile against both cholinesterases. Finally, the introduction of a terminal -OH on the C8 aliphatic chain was performed with the aim of enhancing solubility and membrane permeability. This modification might also allow the establishment of an additional H-bond interaction that potentially increases potency towards the target(s). Collectively, the set of cardanol derivatives
49–
58 was designed with the goal of obtaining cholinesterase inhibitors with concomitant antioxidant and anti-amyloid properties (
Figure 4)
[43].
Figure 4. Cardanol-derived multifunctional compounds 49–58.
The synthesized compounds 49–58 were tested in vitro for their ability to inhibit human AChE and BChE, using the Ellman assay. Then, their anti-amyloid properties were assesed by transmission electron microscopy and their antioxidant (HORAC) profiles were evaluated in comparison with ferulic acid as reference compound. The antioxidant activity was further confirmed in SHSY-5Y cells. Lastly, the assessment of preliminary drug-like properties—in terms of BBB permeation (PAMPA-BBB assay), toxicity in HepG2 cells, plasma stability and kinetic solubility-, was also performed.
The most interesting results are summarized in
Table 4 [43]. As regards to the initial drug-likeness evaluation, they were found potentially BBB permeable, devoid of hepatotoxicity, stable in plasma and soluble. Notably,
52 and
58, which combine cholinesterase activity and anti-amyloid/antioxidant capacity together with good drug-like properties, emerged as the best performing compounds. Thus, we succeeded in identifying phenols
52 and
58 as promising multifunctional cholinesterase inhibitors, modulating amyloid and oxidative cascades.
Table 4. Inhibitory activity against AChE and BChE and antioxidant activity of cardanol derivatives 49–58.
 |
| Compounds |
% Inhibition
hAChE a |
IC50 hAChE
(μM) ± SEM a |
% Inhibition
hBChE a |
IC50 hBChE
(μM) ± SEM a |
HORAC b |
Gallic Acid
Equivalents |
| 49 |
19.4 ± 7.1 |
ND |
68.3 ± 1.3 |
6.74 ± 0.7 |
3.70 ± 0.05 |
| 50 |
12.4 ± 1.2 |
ND |
50.5 ±0.7 |
17.5 ± 3.5 |
5.49 ± 0.58 |
| 51 |
31.1 ± 2.8 |
47.6 ± 4.1 |
59.0 ± 0.8 |
13.3 ± 0.5 |
4.88 ± 0.05 |
| 52 |
40.5 ± 1.8 |
30.0 ± 2.6 |
73.5 ± 0.4 |
6.12 ± 0.8 |
4.37 ± 0.54 |
| 53 |
12.1 ± 0.8 |
ND |
16.6 ± 2.4 |
ND |
4.38 ± 0.23 |
| 54 |
<5 |
ND |
<10 |
ND |
9.73 ± 0.86 |
| 55 |
<10 |
ND |
17.7 ± 3.1 |
ND |
1.49 ± 0.20 |
| 56 |
<5 |
ND |
10.6 ± 2.1 |
ND |
4.80 ± 0.49 |
| 57 |
<10 |
ND |
<5 |
ND |
4.52 ± 0.64 |
| 58 |
<10 |
785 ± 42 |
77.1 ± 0.2 |
4.62 ± 0.14 |
3.50 ± 0,23 |
| 47 |
|
5.65 ± 0.48 |
|
|
n.a. |
| Ferulic acid |
|
|
|
|
4.04 ± 0.51 |
a IC50 inhibitory concentration (μM) or % inhibition at 20 μM of human recombinant AChE and human serum BChE. IC50 values are expressed as mean ± standard error of the mean (SEM) of at least two experiments each performed in triplicate.
b Antioxidant activity is expressed as Gallic Acid Equivalent (GAE). GAE values are expressed as mean ± standard deviation (SD) of three experiments (n = 3). ND = not determined; n.a. = not applicable. Data taken from reference
[43].
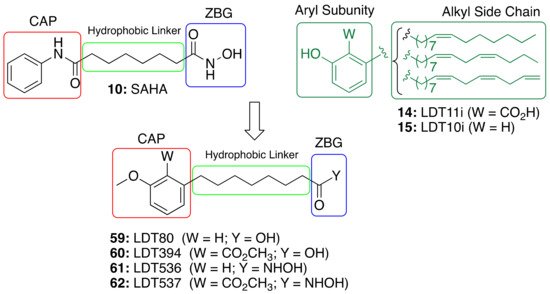
2.4. CNSL-Derived HDAC Inhibitors
The role of saturated anacardic acid
14a as an epigenetic modulator, inhibiting p300 and GCN5 histone acetyltransferases (HAT), is well known
[44]. However,
14a was never reported as a HDAC inhibitor. Given the structural similarity between the hydroxamic HDAC inhibitor vorinostat (
10, SAHA) and the acids LDT80 (
59) and LDT394 (
60), we considered them valuable starting points in the search for CNSL-derived HDAC inhibitors
[45]. The aromatic rings present in the original mixtures of anacardic acids (
14) and cardanols (
15) transformed into O-methylated derivatives represent the CAP subunit—a variation aimed at establishing SAR—while the zinc binding group (ZBG) relies on the hydroxamic acid
61 and
62 generated by the interconversion of the carboxylic acid groups
59 and
60. The hydrophobic linker between CAP and ZBG subunits, found in SAHA, is resembled by a C8 aliphatic chain, characteristic of CNSL derivatives after oxidative cleavage (
Figure 5).
Figure 5. Design of CNSL-derived HDAC inhibitors LDT536 (61) and LDT537 (62) from the octanoic acids LDT80 (59) and LDT394 (60).
The inhibitory profiles of compounds LDT536 (
61) and LDT537 (
62) were assessed against human histone deacetylases of class I (HDAC1, HDAC2, HDAC3 and HDAC8), class II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10) and class IV (HDAC11)
[45]. The obtained results confirmed our rationale, providing CNSL derivatives with a promising HDAC inhibitory profile. Particularly, derivatives
61 and
62 acted mainly on class I and II HDACs, with an inhibition percentage of approximately 80%. As expected,
9 modulated the activity of HDAC1 and 6 with roughly equivalent submicromolar potency. Both
61 and
62 exhibited a similar HDAC inhibitory profile to
9, with inhibitory potencies that were only slightly decreased (by 2.7 to 6.5-fold) (
Table 5). Thus, the obtained results supported the starting idea that the insertion of the hydroxamate moiety on a CNSL backbone provides SAHA-like HDAC inhibitors. Indeed, acid
61, devoid of the hydroxamate moiety, showed only a modest inhibition (12%) at 10 µM (
Table 4). This was further confirmed by Western blots of acetyl-H3, and total H3 after treatment of N9 cells. Additionally, compounds
61 and
62 were able to cross the BBB by passive diffusion as their Pe values were superior to those of standard AD drugs (tacrine, donepezil, and rivastigmine). Moreover, both compounds, and particularly
62, were able to effectively modulate glial cell-induced inflammation and to revert the M1 pro-inflammatory phenotype.
Table 5. In vitro inhibition of HDAC1 and HDAC6 by 60–62 and reference compound (vorinostat).
| Compound |
HDAC1
IC50 ± SEM [nM] |
HDAC6
IC50 ± SEM [nM] |
| 9 |
Vorinostat |
119 |
53 |
| 60 |
LDT394 |
12% * |
12% * |
| 61 |
LDT536 |
316.2 ± 37 |
190.1 ± 38.2 |
| 62 |
LDT537 |
774.7 ± 14.4 |
215.4 ± 28.6 |
* Inhibition at 10 µM. Reprinted with permission from reference
[45], Copyright 2019, American Chemical Society.
2.5. Anti-Inflammatory Activity of Phenolic Lipids from CNSL
As stated above, CNSL phenolic lipids possess strong anti-inflammatory action, which is beneficial for the development of effective anti-AD compounds
[44]. Generally, inflammation can be reduced via the inhibition of pro-inflammatory mediators, as well as the increase of the anti-inflammatory mediator production.
Recently, the anti-inflammatory potential of anacardic acid
14a was studied on lipopolysaccharide (LPS)-induced inflammation in RAW264.7 cell line
[46]. In particular,
14a at 50 µM effectively decreased the expression of the TNF-α, COX-2, iNOS, NF-κB, IL-1β and IL-6 pro-inflammatory genes. Furthermore,
14a was able also to exert protective effects by reducing nitric oxide (NO) and IL-6 production
[46]. Therefore,
14a preventing both the release of pro-inflammatory mediators and downregulating the expression of cytokines emerges as a valuable treatment for preventing inflammation.
The in vitro anti-inflammatory profile was further validated by in vivo studies in Swiss albino mice
[47]. Results from this study revealed that
14a at 25 mg/kg inhibits carrageenan-induced edema. Moreover,
14a decreased leukocyte and neutrophilic migration, and increased the levels of reduced glutathione. It also showed anti-nociceptive activities, by decreasing licking, abdominal writhing, and latency to thermal stimulation. Collectively, these results suggest that
14a possesses anti-inflammatory and antinociceptive properties and also diminishes oxidative stress in acute experimental models.
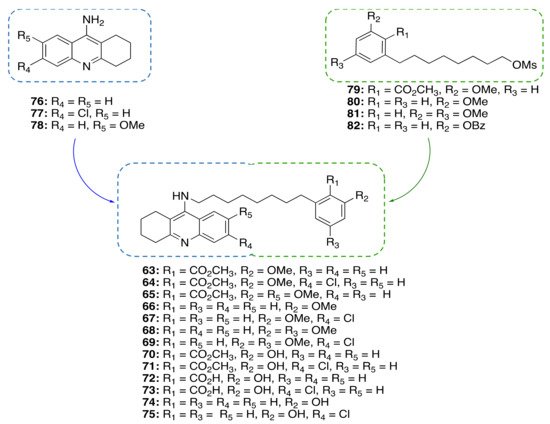
A Framework Combination Approach towards Rationally Designed CNSL-Derived Multitarget Compounds
Based on the promising anti-inflammatory activity of
14a, we applied a framework combination strategy to the design and synthesis of a series of CNSL-derived compounds-tacrine hybrids (
63–75) for the treatment of AD
[48]. We combined in a single chemical entity tacrine moieties (
76–7
8,
Figure 6) with a modified CNSL framework (
79–82). Considering that the presence of the long alkyl chain (C15) of
14a might limit drug-likeness due to excessive lipophilicity, we turned our attention to the shorter-chain (C8) CNSL derivatives (
76–79). In this way, we aimed to create new CNSL-tacrine hybrids which in principle maintained the tacrines’ anticholinesterase activity, combined with the anti-inflammatory activity of CNSL compounds, and possessed drug-like properties. Notably, screening of anticholinesterase activity highlighted potent and selective AChE/BChE inhibitors (
63,
64 and
70), with subnanomolar activities. The excellent BChE activity of
63 (IC50 = 0.0352 nM) was rationalized by the solved X-ray crystal structure. Compounds
63 and
64 exerted neuroprotective/neuroinflammatory effects in microglial BV-2 cells treated with LPS. Particularly,
63 and
64 reduce pro-inflammatory mediators, i.e., TNF-α, IL-1β, iNOS, and COX-2. Furthermore, they acted as anti-neuroinflammatory compounds by inhibiting the transcriptional activation of NF-κB, without causing cytotoxicity in microglial, neuronal, and hepatic cell lines. Lastly, their BBB permeability was also confirmed by BBB-PAMPA assay. All in all, the collected data corroborated our rational design for obtaining effective multitarget anti-AD compounds based on CNSL structure.
Figure 6. Design of CNSL-derived tacrine hybrid compounds 63–75.
 +1 point
+1 point