 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Saima Usman | + 2228 word(s) | 2228 | 2021-10-11 06:06:19 | | | |
| 2 | Jessie Wu | Meta information modification | 2228 | 2021-10-22 09:42:10 | | |
Video Upload Options
Epithelial-mesenchymal transition (EMT) is a reversible plethora of molecular events where epithelial cells gain the phenotype of mesenchymal cells to invade the surrounding tissues. EMT is a physiological event during embryogenesis (type I) but also happens during fibrosis (type II) and cancer metastasis (type III). It is a multifaceted phenomenon governed by the activation of genes associated with cell migration, extracellular matrix degradation, DNA repair, and angiogenesis. The cancer cells employ EMT to acquire the ability to migrate, resist therapeutic agents and escape immunity. One of the key biomarkers of EMT is vimentin, a type III intermediate filament that is normally expressed in mesenchymal cells but is upregulated during cancer metastasis.
1. Introduction
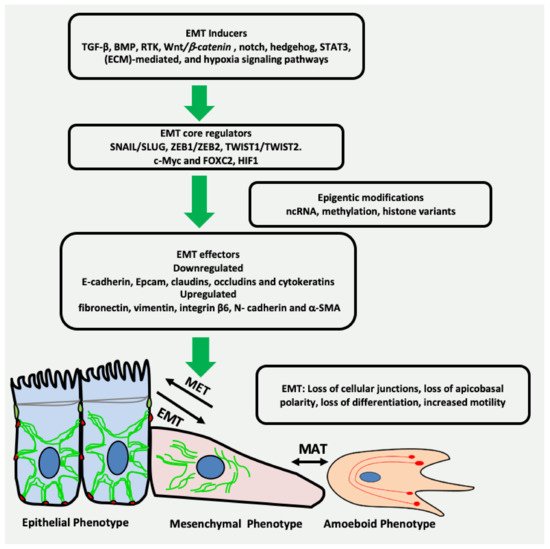
2. EMT May Produce Cancer Stem Cells (CSCs) Expressing Vimentin
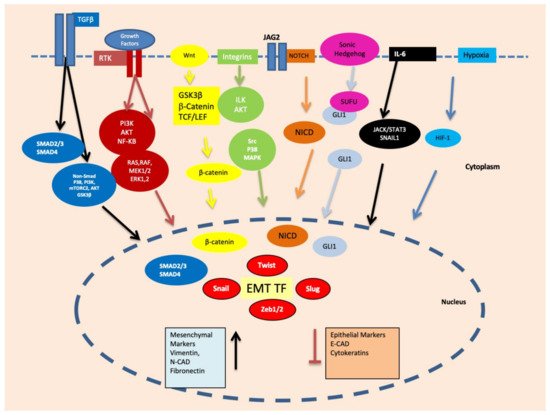
3. Vimentin Expression during Mesenchymal–Amoeboid Transition (MAT) of CSC
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 1–10.
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52.
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776.
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45.
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873.
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018, 194, 161–184.
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2018, 29, 212–226.
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206.
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5511947/ (accessed on 2 October 2021).
- Gonzalez, D.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8.
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial–Mesenchymal Transition. Front. Oncol. 2014, 4, 358.
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499.
- Lin, Y.-T.; Wu, K.-J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 1–10.
- Scanlon, C.; Van Tubergen, E.; Inglehart, R.; D’Silva, N. Biomarkers of Epithelial-Mesenchymal Transition in Squamous Cell Carcinoma. J. Dent. Res. 2012, 92, 114–121.
- Danielsson, F.; Peterson, M.K.; Araújo, H.C.; Lautenschläger, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147.
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 1–9.
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983.
- Ivaska, J. Vimentin. Small GTPases 2011, 2, 51–53.
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.-P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2010, 30, 1436–1448.
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092.
- Pattabiraman, S.; Azad, G.K.; Amen, T.; Brielle, S.; Park, J.E.; Sze, S.K.; Meshorer, E.; Kaganovich, D. Vimentin protects differentiating stem cells from stress. Sci. Rep. 2020, 10, 1–15.
- Usman, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Major Molecular Signaling Pathways in Oral Cancer Associated With Therapeutic Resistance. Front. Oral Heal. 2021, 1, 15.
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity – A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79.
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. BioEssays 2016, 38, S65–S74.
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Ann. Rev. Pathol. Mech. Dis. 2014, 9, 47–71.
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668.
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940.
- Scheel, C.; Weinberg, R.A. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 2011, 129, 2310–2314.
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280.
- Zhang, R.; Tu, J.; Liu, S. Novel molecular regulators of breast cancer stem cell plasticity and heterogeneity. Semin. Cancer Biol. 2021.
- Zhang, X.; Zhao, W.; Li, Y. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95.
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151.
- Thapa, R.; Wilson, G. The Importance of CD44 as a Stem Cell Biomarker and Therapeutic Target in Cancer. Stem Cells Int. 2016, 2016, 1–15.
- Patteson, A.E.; Vahabikashi, A.; Goldman, R.D.; Janmey, P.A. Mechanical and Non-Mechanical Functions of Filamentous and Non-Filamentous Vimentin. BioEssays 2020, 42, e2000078.
- Fang, X.; Zheng, P.; Tang, J.; Liu, Y. CD24: From A to Z. Cell. Mol. Immunol. 2010, 7, 100–103.
- Meyer, M.J.; Fleming, J.M.; Ali, M.A.; Pesesky, M.W.; Ginsburg, E.; Vonderhaar, B.K. Dynamic regulation of CD24 and the invasive, CD44posCD24negphenotype in breast cancer cell lines. Breast Cancer Res. 2009, 11, 14–82.
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 2007, 1, 555–567.
- Pearce, D.J.; Taussig, D.; Simpson, C.; Allen, K.; Rohatiner, A.Z.; Lister, T.A.; Bonnet, D. Characterization of Cells with a High Aldehyde Dehydrogenase Activity from Cord Blood and Acute Myeloid Leukemia Samples. Stem Cells 2005, 23, 752–760.
- Wu, A.; Luo, W.; Zhang, Q.; Yang, Z.; Zhang, G.; Li, S.; Yao, K. Aldehyde dehydrogenase 1, a functional marker for identifying cancer stem cells in human nasopharyngeal carcinoma. Cancer Lett. 2013, 330, 181–189.
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2013, 2, 78–91.
- Wang, J.; Zeng, H.; Li, H.; Zhang, J.; Wang, S. Roles of sex-determining region Y-box 2 in cell pluripotency and tumor-related signaling pathways. Mol. Clin. Oncol. 2015, 3, 1203–1207.
- Li, B.; Chen, M.; Pan, M.-X. Sex determining region Y-box 2 is a prognostic factor for head and neck squamous cell carcinoma: Evidence from 11 published investigations. J. Cancer Res. Ther. 2020, 16, 434.
- Freier, K.; Knoepfle, K.; Flechtenmacher, C.; Pungs, S.; Devens, F.; Toedt, G.; Hofele, C.; Joos, S.; Lichter, P.; Radlwimmer, B. Recurrent copy number gain of transcription factorSOX2and corresponding high protein expression in oral squamous cell carcinoma. Genes Chromosom. Cancer 2010, 49, 9–16.
- Bayo, P.; Jou, A.; Stenzinger, A.; Shao, C.; Gross, M.; Jensen, A.D.; Grabe, N.; Mende, C.H.; Rados, P.V.; Debus, J.; et al. Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Mol. Oncol. 2015, 9, 1704–1719.
- Deng, Z.; Du, W.W.; Fang, L.; Shan, S.W.; Qian, J.; Lin, J.; Qian, W.; Ma, J.; Rutnam, Z.J.; Yang, B.B. The Intermediate Filament Vimentin Mediates MicroRNA miR-378 Function in Cellular Self-renewal by Regulating the Expression of the Sox2 Transcription Factor*. J. Biol. Chem. 2013, 288, 319–331.
- Waldeyer, R. Die Entwicklung der Carcinome. Virchows Arch. F. Path. Anat. 1872, 55, 67–159.
- Waldeyer., R. Die Entwicklung der Carcinome. Virchows Arch. F. Path. Anat. 1867, 41, 470–523.
- Enterline, H.T.; Coman, D.R. The ameboid motility of human and animal neoplastic cells. Cancer 1950, 3, 1033–1038.
- Taddei, M.L.; Giannoni, E.; Morandi, A.; Ippolito, L.; Ramazzotti, M.; Callari, M.; Gandellini, P.; Chiarugi, P. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun. Signal. 2014, 12, 24.
- Emad, A.; Ray, T.; Jensen, T.; Parat, M.; Natrajan, R.; Sinha, S.; Ray, S.P. An epithelial-mesenchymal-amoeboid transition gene signature reveals subtypes of breast cancer progression and metastasis. bioRxiv 2017. Available online: https://www.biorxiv.org/content/10.1101/219410v2.full (accessed on 13 September 2021).
- Wu, J.-S.; Jiang, J.; Chen, B.-J.; Wang, K.; Tang, Y.-L.; Liang, X.-H. Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 2020, 14, 100899.
- Krakhmal, N.V.; Zavyalova, M.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V. Cancer Invasion: Patterns and Mechanisms. Acta Naturae 2015, 7, 17–28.
- Talkenberger, K.; Cavalcanti-Adam, E.; Voss-Böhme, A.; Deutsch, A. Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments. Sci. Rep. 2017, 7, 1–12.
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752.
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Bröcker, E.-B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277.
- Holle, A.; Devi, N.G.K.; Clar, K.; Fan, A.; Saif, M.T.; Kemkemer, R.; Spatz, J.P. Cancer Cells Invade Confined Microchannels via a Self-Directed Mesenchymal-to-Amoeboid Transition. Nano Lett. 2019, 19, 2280–2290.
- Lavenus, S.B.; Tudor, S.M.; Ullo, M.F.; Vosatka, K.W.; Logue, J.S. A flexible network of vimentin intermediate filaments promotes migration of amoeboid cancer cells through confined environments. J. Biol. Chem. 2020, 295, 6700–6709.
- Strouhalova, K.; Přechová, M.; Gandalovičová, A.; Brábek, J.; Gregor, M.; Rosel, D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers 2020, 12, 184.
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 1–36.
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769.
- Melzer, C.; Von Der Ohe, J.; Hass, R. Breast Carcinoma: From Initial Tumor Cell Detachment to Settlement at Secondary Sites. BioMed Res. Int. 2017, 2017, 1–11.



