 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Virinder Kaur Sarhadi | + 4215 word(s) | 4215 | 2021-10-18 10:34:59 | | | |
| 2 | Vivi Li | Meta information modification | 4215 | 2021-10-21 10:07:33 | | |
Video Upload Options
Osteosarcoma (OS) is an aggressive bone tumor that mainly affects children and adolescents. OS has a strong tendency to relapse and metastasize, resulting in poor prognosis and survival. The high heterogeneity and genetic complexity of OS make it challenging to identify new therapeutic targets. Mesenchymal stem cells (MSCs) are multipotent stem cells that can differentiate into adipocytes, osteoblasts, or chondroblasts. OS is thought to originate at some stage in the differentiation process of MSC to pre-osteoblast or from osteoblast precursors. MSCs contribute to OS progression by interacting with tumor cells via paracrine signaling and affect tumor cell proliferation, invasion, angiogenesis, immune response, and metastasis. Extracellular vesicles (EVs), secreted by OS cells and MSCs in the tumor microenvironment, are crucial mediators of intercellular communication, driving OS progression by transferring miRNAs/RNA and proteins to other cells. MSC-derived EVs have both pro-tumor and anti-tumor effects on OS progression. MSC-EVs can be also engineered to deliver anti-tumor cargo to the tumor site, which offers potential applications in MSC-EV-based OS treatment.
1. Introduction
2. Mesenchymal Stem Cells’ Role in Osteosarcoma
2.1. Cellular Origin of OS
2.2. MSCs in OS Microenvironment
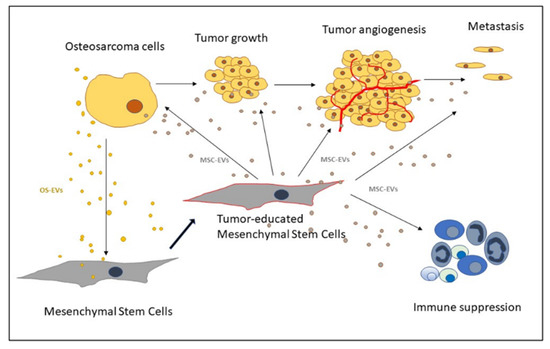
2.3. Recruitment of MSCs to the Tumor Site
2.4. OS Microenvironment-Induced Changes in MSCs
| OS Signal | Effect on MSCs | MSC Signal | Effect on OS | Reference |
|---|---|---|---|---|
| Exosomes | Increase in COLGALT2 and proliferation. | Wang et al. 2020 [28] | ||
| CM 1/co-culture | Increase in MMP2/9; STAT3 activation. Increased proliferation, invasion, and metastasis. | Wang et al. 2017 [29] | ||
| IL-8 (co-culture) |
Increased IL-8. | IL-8 (co-culture) |
Increased IL-8. Increased metastatic potential. |
Kawano et al. 2018 [30] |
| IL-6 CM/co-culture |
Increase in MMP2/9; JAK2/STAT3 activation. Increased proliferation, migration, and doxorubicin resistance. | Lu et al. 2021 [31] | ||
| MCP-1, GRO-α, and TGFβ | Mesenchymal-to-amoeboid transition. Increase in MCP-1, GRO-α, IL-6, and IL-8 in the tumor environment. |
Increased migration, invasion, and trans-endothelial migration. | Pietrovito et al. 2018 [20] |
|
| OS-EVs | LINE-1 hypomethylation increased VEGF-A. | Mannerström et al. 2019 [24] | ||
| CM | STAT3 activation. Promote survival and drug resistance. |
Tu et al. 2016 [25] | ||
| MSC-EVs (under stress) |
Increased migration. Apoptosis resistance. | Vallabhaneni et al. 2016 [32] | ||
| Co-culture | Increased TGFβ. | Co-culture IL-6 |
Increased OS proliferation, stemness & migration. | Cortini et al. 2016 [26] |
| IL-6 | STAT3 activation. Increased proliferation and metastasis. | Tu et al. 2012 [33] | ||
| CM/TGFβ | Increased IL-6, VEGF. Inhibit osteogenic differentiation. |
Tu et al. 2014 [34] | ||
| IL-8 | CXCR1/Akt activation. Promotes metastasis. |
Du et al. 2018 [35] | ||
| EVs/TGFβ | Increased IL-6. | Tumor-educated MSC | Activation of STAT3 signaling. | Baglio et al. 2017 [23] |
| EVs | Cell growth under hypoxia. Activation of PI3K/AKT & HIF-1α. | Lin et al. 2019 [36] | ||
| EVs | Activation of Hedgehog signaling. Tumor growth. | Qi et al. 2017 [37] |
2.5. Transformation of MSCs into Carcinoma-Associated Fibroblasts
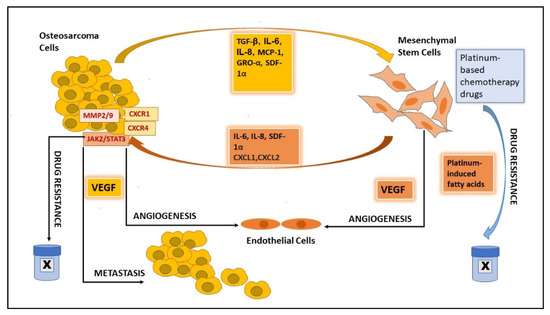
2.6. MSCs’ Role in Promoting OS Growth and Progression
2.7. MSCs’ Role in Angiogenesis
3. Extracellular Vesicles as Mediators of MSCs and Osteosarcoma Crosstalk
3.1. OS-EVs
| Source of EVs | miRNAs/other RNAs in EVs | Proteins in EVs | Function | Reference |
|---|---|---|---|---|
| OS cells | miR-146a-5p, miR-10b-5p, miR-143-3p, miR-382-5p, miR-150-5p, miR-125b-5p, miR-27a-3p, miR-145-5p, miR-26a-5p, miR-93-5p, miR-21-5p, miR-92a-3p, and miR-106a-5p | serpin-E1, serpin-F1, TIMP-1, thrombospondin-1, urokinase-type plasminogen activator (uPA), VEGF, pentraxin-3, PDGF-AA, angiopoietin-2, coagulation factor-III, CD26, CD105, endostatin, endothelin-1, and HB-EGF | Angiogenesis. | Perut et al. 2019 [53] |
| OS cells & tissue | lncRNA OIP5-AS1 | Angiogenesis. | Li et al. 2021 [54] | |
| OS cells | miR-148a-3p and miR-21-5p | TME remodeling. | Raimondi et al. 2020 [49] | |
| OS cells | TGFβ | Increase IL6 in AD-MSCs, tumor growth, STAT3 activation, and lung metastasis. Autophagy. |
Baglio et al. 2017, Tu et al. 2012, Huang et al. 2020 [23][33][55] | |
| BM-MSC | lncRNA MALAT1 | Proliferation, invasion, and migration of OS cells via lncRNA MALAT1/miR-143/NRSN2/Wnt/β-Catenin Axis. | Li et al. 2021 [56] | |
| BM-MSC | non-coding RNA PVT1 | OS migration by upregulating ERG and sponging miR-183-5p in OS cells. | Zhao et al. 2019 [57] | |
| BM-MSC | miR-206 | Tumor suppression and apoptosis. | Zhang et al. 2020 [58] | |
| BM-MSC | microRNA-208a LCP1 |
OS cell migration & invasion. OS proliferation and metastasis via the JAK2/STAT3 pathway. |
Qin et al. 2020 [59] | |
| Highly metastatic OS cells | NPM1, CCT2, CCT4, CCT6A, CCT8, VIM, CLTC, COL6A2, HNRNPC, PKM, ACTN4, MYH10, PAICS, VCP, ANXA1, ACLY | Metastasis. | Macklin et al. 2016 [60] | |
| Engineered AD-MSC | miR-101; miR-150 synthetic miR-143 |
OS Therapy. Suppress OS growth. | Zhang et al. 2020; Shimbo et al. 2014; Xu et al. 2020; [61][62] |
3.2. MSC-EVs
3.3. MSC-EVs in Angiogenesis
3.4. MSC-EVs in OS Metastasis
3.5. MSC-EVs in Immune Response
References
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma Incidence and Survival Rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543.
- ESMO European Sarcoma Network Working Group. Bone Sarcomas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2012, 23, vii100-9.
- Broadhead, M.L.; Clark, J.C.; Myers, D.E.; Dass, C.R.; Choong, P.F. The Molecular Pathogenesis of Osteosarcoma: A Review. Sarcoma 2011, 2011, 959248.
- Kempf-Bielack, B.; Bielack, S.S.; Jurgens, H.; Branscheid, D.; Berdel, W.E.; Exner, G.U.; Gobel, U.; Helmke, K.; Jundt, G.; Kabisch, H.; et al. Osteosarcoma Relapse After Combined Modality Therapy: An Analysis of Unselected Patients in the Cooperative Osteosarcoma Study Group (COSS). J. Clin. Oncol. 2005, 23, 559–568.
- Bhaskar, B.; Mekala, N.K.; Baadhe, R.R.; Rao, P.S. Role of Signaling Pathways in Mesenchymal Stem Cell Differentiation. Curr. Stem Cell. Res. Ther. 2014, 9, 508–512.
- Main, H.; Munsie, M.; O’Connor, M.D. Managing the Potential and Pitfalls during Clinical Translation of Emerging Stem Cell Therapies. Clin. Transl. Med. 2014, 3, 1–10.
- Yang, Y.; Yang, R.; Roth, M.; Piperdi, S.; Zhang, W.; Dorfman, H.; Rao, P.; Park, A.; Tripathi, S.; Freeman, C.; et al. Genetically Transforming Human Osteoblasts to Sarcoma: Development of an Osteosarcoma Model. Genes Cancer 2017, 8, 484–494.
- Rubio, R.; Gutierrez-Aranda, I.; Saez-Castillo, A.I.; Labarga, A.; Rosu-Myles, M.; Gonzalez-Garcia, S.; Toribio, M.L.; Menendez, P.; Rodriguez, R. The Differentiation Stage of p53-Rb-Deficient Bone Marrow Mesenchymal Stem Cells Imposes the Phenotype of in vivo Sarcoma Development. Oncogene 2013, 32, 4970–4980.
- Mutsaers, A.J.; Walkley, C.R. Cells of Origin in Osteosarcoma: Mesenchymal Stem Cells Or Osteoblast Committed Cells? Bone 2014, 62, 56–63.
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal Stem Cells in Health and Disease. Nat. Rev. Immunol. 2008, 8, 726–736.
- Jo, V.Y.; Fletcher, C.D. WHO Classification of Soft Tissue Tumours: An Update Based on the 2013 (4th) Edition. Pathology 2014, 46, 95–104.
- Mohseny, A.B.; Szuhai, K.; Romeo, S.; Buddingh, E.P.; Briaire-de Bruijn, I.; de Jong, D.; van Pel, M.; Cleton-Jansen, A.M.; Hogendoorn, P.C. Osteosarcoma Originates from Mesenchymal Stem Cells in Consequence of Aneuploidization and Genomic Loss of Cdkn2. J. Pathol. 2009, 219, 294–305.
- Wang, J.Y.; Wu, P.K.; Chen, P.C.; Lee, C.W.; Chen, W.M.; Hung, S.C. Generation of Osteosarcomas from a Combination of Rb Silencing and C-Myc Overexpression in Human Mesenchymal Stem Cells. Stem Cells Transl. Med. 2017, 6, 512–526.
- Walkley, C.R.; Qudsi, R.; Sankaran, V.G.; Perry, J.A.; Gostissa, M.; Roth, S.I.; Rodda, S.J.; Snay, E.; Dunning, P.; Fahey, F.H.; et al. Conditional Mouse Osteosarcoma, Dependent on p53 Loss and Potentiated by Loss of Rb, Mimics the Human Disease. Genes Dev. 2008, 22, 1662–1676.
- Dani, N.; Olivero, M.; Mareschi, K.; van Duist, M.M.; Miretti, S.; Cuvertino, S.; Patane, S.; Calogero, R.; Ferracini, R.; Scotlandi, K.; et al. The MET Oncogene Transforms Human Primary Bone-Derived Cells into Osteosarcomas by Targeting Committed Osteo-Progenitors. J. Bone Miner. Res. 2012, 27, 1322–1334.
- Chang, X.; Ma, Z.; Zhu, G.; Lu, Y.; Yang, J. New Perspective into Mesenchymal Stem Cells: Molecular Mechanisms Regulating Osteosarcoma. J. Bone Oncol. 2021, 29, 100372.
- Abarrategi, A.; Tornin, J.; Martinez-Cruzado, L.; Hamilton, A.; Martinez-Campos, E.; Rodrigo, J.P.; Gonzalez, M.V.; Baldini, N.; Garcia-Castro, J.; Rodriguez, R. Osteosarcoma: Cells-of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int. 2016, 2016, 3631764.
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem. Cells 2020, 9, 976.
- Sun, Z.; Wang, S.; Zhao, R.C. The Roles of Mesenchymal Stem Cells in Tumor Inflammatory Microenvironment. J. Hematol. Oncol. 2014, 7, 14.
- Pietrovito, L.; Leo, A.; Gori, V.; Lulli, M.; Parri, M.; Becherucci, V.; Piccini, L.; Bambi, F.; Taddei, M.L.; Chiarugi, P. Bone Marrow-Derived Mesenchymal Stem Cells Promote Invasiveness and Transendothelial Migration of Osteosarcoma Cells Via a Mesenchymal to Amoeboid Transition. Mol. Oncol. 2018, 12, 659–676.
- Gutova, M.; Najbauer, J.; Frank, R.T.; Kendall, S.E.; Gevorgyan, A.; Metz, M.Z.; Guevorkian, M.; Edmiston, M.; Zhao, D.; Glackin, C.A.; et al. Urokinase Plasminogen Activator and Urokinase Plasminogen Activator Receptor Mediate Human Stem Cell Tropism to Malignant Solid Tumors. Stem Cells 2008, 26, 1406–1413.
- Ho, I.A.; Yulyana, Y.; Sia, K.C.; Newman, J.P.; Guo, C.M.; Hui, K.M.; Lam, P.Y. Matrix metalloproteinase-1-mediated mesenchymal stem cell tumor tropism is dependent on crosstalk with stromal derived growth factor 1/C-X-C chemokine receptor 4 axis. FASEB J. 2014, 28, 4359–4368.
- Baglio, S.R.; Lagerweij, T.; Perez-Lanzon, M.; Ho, X.D.; Leveille, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer Res. 2017, 23, 3721–3733.
- Mannerstrom, B.; Kornilov, R.; Abu-Shahba, A.G.; Chowdhury, I.M.; Sinha, S.; Seppanen-Kaijansinkko, R.; Kaur, S. Epigenetic Alterations in Mesenchymal Stem Cells by Osteosarcoma-Derived Extracellular Vesicles. Epigenetics 2019, 14, 352–364.
- Tu, B.; Zhu, J.; Liu, S.; Wang, L.; Fan, Q.; Hao, Y.; Fan, C.; Tang, T.T. Mesenchymal Stem Cells Promote Osteosarcoma Cell Survival and Drug Resistance through Activation of STAT3. Oncotarget 2016, 7, 48296–48308.
- Cortini, M.; Massa, A.; Avnet, S.; Bonuccelli, G.; Baldini, N. Tumor-Activated Mesenchymal Stromal Cells Promote Osteosarcoma Stemness and Migratory Potential Via IL-6 Secretion. PLoS ONE 2016, 11, e0166500.
- Avnet, S.; Di Pompo, G.; Chano, T.; Errani, C.; Ibrahim-Hashim, A.; Gillies, R.J.; Donati, D.M.; Baldini, N. Cancer-Associated Mesenchymal Stroma Fosters the Stemness of Osteosarcoma Cells in Response to Intratumoral Acidosis Via NF-kappaB Activation. Int. J. Cancer 2017, 140, 1331–1345.
- Wang, Y.; Chu, Y.; Li, K.; Zhang, G.; Guo, Z.; Wu, X.; Qiu, C.; Li, Y.; Wan, X.; Sui, J.; et al. Exosomes Secreted by Adipose-Derived Mesenchymal Stem Cells Foster Metastasis and Osteosarcoma Proliferation by Increasing COLGALT2 Expression. Front. Cell. Dev. Biol. 2020, 8, 353.
- Wang, Y.; Chu, Y.; Yue, B.; Ma, X.; Zhang, G.; Xiang, H.; Liu, Y.; Wang, T.; Wu, X.; Chen, B. Adipose-Derived Mesenchymal Stem Cells Promote Osteosarcoma Proliferation and Metastasis by Activating the STAT3 Pathway. Oncotarget 2017, 8, 23803–23816.
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Tsumura, H. Interaction between Human Osteosarcoma and Mesenchymal Stem Cells Via an Interleukin-8 Signaling Loop in the Tumor Microenvironment. Cell. Commun. Signal. 2018, 16, 13.
- Lu, M.; Xie, K.; Lu, X.; Lu, L.; Shi, Y.; Tang, Y. Notoginsenoside R1 Counteracts Mesenchymal Stem Cell-Evoked Oncogenesis and Doxorubicin Resistance in Osteosarcoma Cells by Blocking IL-6 Secretion-Induced JAK2/STAT3 Signaling. Invest. New Drugs 2021, 39, 416–425.
- Vallabhaneni, K.C.; Hassler, M.Y.; Abraham, A.; Whitt, J.; Mo, Y.Y.; Atfi, A.; Pochampally, R. Mesenchymal Stem/Stromal Cells Under Stress Increase Osteosarcoma Migration and Apoptosis Resistance Via Extracellular Vesicle Mediated Communication. PLoS ONE 2016, 11, e0166027.
- Tu, B.; Du, L.; Fan, Q.M.; Tang, Z.; Tang, T.T. STAT3 Activation by IL-6 from Mesenchymal Stem Cells Promotes the Proliferation and Metastasis of Osteosarcoma. Cancer Lett. 2012, 325, 80–88.
- Tu, B.; Peng, Z.X.; Fan, Q.M.; Du, L.; Yan, W.; Tang, T.T. Osteosarcoma Cells Promote the Production of Pro-Tumor Cytokines in Mesenchymal Stem Cells by Inhibiting their Osteogenic Differentiation through the TGF-Beta/Smad2/3 Pathway. Exp. Cell Res. 2014, 320, 164–173.
- Du, L.; Han, X.G.; Tu, B.; Wang, M.Q.; Qiao, H.; Zhang, S.H.; Fan, Q.M.; Tang, T.T. CXCR1/Akt Signaling Activation Induced by Mesenchymal Stem Cell-Derived IL-8 Promotes Osteosarcoma Cell Anoikis Resistance and Pulmonary Metastasis. Cell. Death Dis. 2018, 9, 714.
- Lin, S.; Zhu, B.; Huang, G.; Zeng, Q.; Wang, C. Microvesicles Derived from Human Bone Marrow Mesenchymal Stem Cells Promote U2OS Cell Growth Under Hypoxia: The Role of PI3K/AKT and HIF-1alpha. Hum. Cell 2019, 32, 64–74.
- Qi, J.; Zhou, Y.; Jiao, Z.; Wang, X.; Zhao, Y.; Li, Y.; Chen, H.; Yang, L.; Zhu, H.; Li, Y. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth through Hedgehog Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2242–2254.
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell. Dev. Biol. 2019, 7, 60.
- Zhu, H.; Guo, S.; Zhang, Y.; Yin, J.; Yin, W.; Tao, S.; Wang, Y.; Zhang, C. Proton-Sensing GPCR-YAP Signalling Promotes Cancer-Associated Fibroblast Activation of Mesenchymal Stem Cells. Int. J. Biol. Sci. 2016, 12, 389–396.
- Lin, L.; Huang, K.; Guo, W.; Zhou, C.; Wang, G.; Zhao, Q. Conditioned Medium of the Osteosarcoma Cell Line U2OS Induces hBMSCs to Exhibit Characteristics of Carcinoma-Associated Fibroblasts Via Activation of IL-6/STAT3 Signalling. J. Biochem. 2020, 168, 265–271.
- Wang, Y.M.; Wang, W.; Qiu, E.D. Osteosarcoma Cells Induce Differentiation of Mesenchymal Stem Cells into Cancer Associated Fibroblasts through Notch and Akt Signaling Pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 8479–8486.
- Maurizi, G.; Verma, N.; Gadi, A.; Mansukhani, A.; Basilico, C. Sox2 is Required for Tumor Development and Cancer Cell Proliferation in Osteosarcoma. Oncogene 2018, 37, 4626–4632.
- Xu, W.T.; Bian, Z.Y.; Fan, Q.M.; Li, G.; Tang, T.T. Human Mesenchymal Stem Cells (hMSCs) Target Osteosarcoma and Promote its Growth and Pulmonary Metastasis. Cancer Lett. 2009, 281, 32–41.
- Zhou, S. TGF-Beta Regulates Beta-Catenin Signaling and Osteoblast Differentiation in Human Mesenchymal Stem Cells. J. Cell. Biochem. 2011, 112, 1651–1660.
- Li, G.C.; Zhang, H.W.; Zhao, Q.C.; Sun, L.I.; Yang, J.J.; Hong, L.; Feng, F.; Cai, L. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor β1. Oncol. Lett. 2016, 11, 1089–1094.
- Batlle, R.; Andrés, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38α through TGF-β and JNK signaling. Nat. Commun. 2019, 10, 3071.
- Chaturvedi, P.; Gilkes, D.M.; Wong, C.C.; Kshitiz, J.; Luo, W.; Zhang, H.; Wei, H.; Takano, N.; Schito, L.; Levchenko, A.; et al. Hypoxia-Inducible Factor-Dependent Breast Cancer-Mesenchymal Stem Cell Bidirectional Signaling Promotes Metastasis. J. Clin. Invest. 2013, 123, 189–205.
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, 977.
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma Cell-Derived Exosomes Affect Tumor Microenvironment by Specific Packaging of microRNAs. Carcinogenesis 2020, 41, 666–677.
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360.
- Lobb, R.J.; Lima, L.G.; Moller, A. Exosomes: Key Mediators of Metastasis and Pre-Metastatic Niche Formation. Semin. Cell Dev. Biol. 2017, 67, 3–10.
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-Secreted miR-9 Promotes Endothelial Cell Migration and Angiogenesis by Activating the JAK-STAT Pathway. EMBO J. 2012, 31, 3513–3523.
- Perut, F.; Roncuzzi, L.; Zini, N.; Massa, A.; Baldini, N. Extracellular Nanovesicles Secreted by Human Osteosarcoma Cells Promote Angiogenesis. Cancers 2019, 11, 779.
- Li, Y.; Lin, S.; Xie, X.; Zhu, H.; Fan, T.; Wang, S. Highly Enriched Exosomal lncRNA OIP5-AS1 Regulates Osteosarcoma Tumor Angiogenesis and Autophagy through miR-153 and ATG5. Am. J. Transl. Res. 2021, 13, 4211–4223.
- Huang, Y.; Liu, W.; He, B.; Wang, L.; Zhang, F.; Shu, H.; Sun, L. Exosomes Derived from Bone Marrow Mesenchymal Stem Cells Promote Osteosarcoma Development by Activating Oncogenic Autophagy. J. Bone Oncol. 2020, 21, 100280.
- Li, F.; Chen, X.; Shang, C.; Ying, Q.; Zhou, X.; Zhu, R.; Lu, H.; Hao, X.; Dong, Q.; Jiang, Z. Bone Marrow Mesenchymal Stem Cells-Derived Extracellular Vesicles Promote Proliferation, Invasion and Migration of Osteosarcoma Cells via the lncRNA MALAT1/miR-143/NRSN2/Wnt/Beta-Catenin Axis. Onco Targets Ther. 2021, 14, 737–749.
- Zhao, W.; Qin, P.; Zhang, D.; Cui, X.; Gao, J.; Yu, Z.; Chai, Y.; Wang, J.; Li, J. Long Non-Coding RNA PVT1 Encapsulated in Bone Marrow Mesenchymal Stem Cell-Derived Exosomes Promotes Osteosarcoma Growth and Metastasis by Stabilizing ERG and Sponging miR-183-5p. Aging 2019, 11, 9581–9596.
- Zhang, H.; Wang, J.; Ren, T.; Huang, Y.; Liang, X.; Yu, Y.; Wang, W.; Niu, J.; Guo, W. Bone Marrow Mesenchymal Stem Cell-Derived Exosomal miR-206 Inhibits Osteosarcoma Progression by Targeting TRA2B. Cancer Lett. 2020, 490, 54–65.
- Qin, F.; Tang, H.; Zhang, Y.; Zhang, Z.; Huang, P.; Zhu, J. Bone Marrow-Derived Mesenchymal Stem Cell-Derived Exosomal microRNA-208a Promotes Osteosarcoma Cell Proliferation, Migration, and Invasion. J. Cell. Physiol. 2020, 235, 4734–4745.
- Macklin, R.; Wang, H.; Loo, D.; Martin, S.; Cumming, A.; Cai, N.; Lane, R.; Ponce, N.S.; Topkas, E.; Inder, K.; et al. Extracellular Vesicles Secreted by Highly Metastatic Clonal Variants of Osteosarcoma Preferentially Localize to the Lungs and Induce Metastatic Behaviour in Poorly Metastatic Clones. Oncotarget 2016, 7, 43570–43587.
- Zhang, K.; Dong, C.; Chen, M.; Yang, T.; Wang, X.; Gao, Y.; Wang, L.; Wen, Y.; Chen, G.; Wang, X.; et al. Extracellular Vesicle-Mediated Delivery of miR-101 Inhibits Lung Metastasis in Osteosarcoma. Theranostics 2020, 10, 411–425.
- Shimbo, K.; Miyaki, S.; Ishitobi, H.; Kato, Y.; Kubo, T.; Shimose, S.; Ochi, M. Exosome-Formed Synthetic microRNA-143 is Transferred to Osteosarcoma Cells and Inhibits their Migration. Biochem. Biophys. Res. Commun. 2014, 445, 381–387.
- Kobayashi, E.; Hornicek, F.J.; Duan, Z. MicroRNA Involvement in Osteosarcoma. Sarcoma 2012, 2012, 359739.
- Alcayaga-Miranda, F.; Varas-Godoy, M.; Khoury, M. Harnessing the Angiogenic Potential of Stem Cell-Derived Exosomes for Vascular Regeneration. Stem Cells Int. 2016, 2016, 3409169.
- Gonzalez-King, H.; Garcia, N.A.; Ontoria-Oviedo, I.; Ciria, M.; Montero, J.A.; Sepulveda, P. Hypoxia Inducible Factor-1alpha Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem Cells 2017, 35, 1747–1759.
- Gong, M.; Yu, B.; Wang, J.; Wang, Y.; Liu, M.; Paul, C.; Millard, R.W.; Xiao, D.S.; Ashraf, M.; Xu, M. Mesenchymal Stem Cells Release Exosomes that Transfer miRNAs to Endothelial Cells and Promote Angiogenesis. Oncotarget 2017, 8, 45200–45212.
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, J.Y.; Kim, C.W. Exosomes Derived from Mesenchymal Stem Cells Suppress Angiogenesis by Down-Regulating VEGF Expression in Breast Cancer Cells. PLoS ONE 2013, 8, e84256.
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth in vivo. Cancer Lett. 2012, 315, 28–37.
- Bian, Z.Y.; Fan, Q.M.; Li, G.; Xu, W.T.; Tang, T.T. Human Mesenchymal Stem Cells Promote Growth of Osteosarcoma: Involvement of Interleukin-6 in the Interaction between Human Mesenchymal Stem Cells and Saos-2. Cancer Sci. 2010, 101, 2554–2560.
- Le Blanc, K.; Ringden, O. Immunobiology of Human Mesenchymal Stem Cells and Future use in Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2005, 11, 321–334.
- Zhang, B.; Yin, Y.; Lai, R.C.; Tan, S.S.; Choo, A.B.; Lim, S.K. Mesenchymal Stem Cells Secrete Immunologically Active Exosomes. Stem Cells Dev. 2014, 23, 1233–1244.
- Mardpour, S.; Hamidieh, A.A.; Taleahmad, S.; Sharifzad, F.; Taghikhani, A.; Baharvand, H. Interaction between Mesenchymal Stromal Cell-Derived Extracellular Vesicles and Immune Cells by Distinct Protein Content. J. Cell. Physiol. 2019, 234, 8249–8258.
- Zhang, Q.; Fu, L.; Liang, Y.; Guo, Z.; Wang, L.; Ma, C.; Wang, H. Exosomes Originating from MSCs Stimulated with TGF-Beta and IFN-Gamma Promote Treg Differentiation. J. Cell. Physiol. 2018, 233, 6832–6840.
- Khare, D.; Or, R.; Resnick, I.; Barkatz, C.; Almogi-Hazan, O.; Avni, B. Mesenchymal Stromal Cell-Derived Exosomes Affect mRNA Expression and Function of B-Lymphocytes. Front. Immunol. 2018, 9, 3053.
- Luo, Z.W.; Liu, P.P.; Wang, Z.X.; Chen, C.Y.; Xie, H. Macrophages in Osteosarcoma Immune Microenvironment: Implications for Immunotherapy. Front. Oncol. 2020, 10, 586580.
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207.
- Showalter, M.R.; Wancewicz, B.; Fiehn, O.; Archard, J.A.; Clayton, S.; Wagner, J.; Deng, P.; Halmai, J.; Fink, K.D.; Bauer, G.; et al. Primed Mesenchymal Stem Cells Package Exosomes with Metabolites Associated with Immunomodulation. Biochem. Biophys. Res. Commun. 2019, 512, 729–735.
- Jia, X.H.; Feng, G.W.; Wang, Z.L.; Du, Y.; Shen, C.; Hui, H.; Peng, D.; Li, Z.J.; Kong, D.L.; Tian, J. Activation of mesenchymal stem cells by macrophages promotes tumor progression through immune suppressive effects. Oncotarget 2016, 12, 20934–20944.
- Shao, X.J.; Xiang, S.F.; Chen, Y.Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.D.; He, Q.J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta. Pharmacol. Sin. 2019, 40, 1343–1350.
- Gunassekaran, G.R.; Poongkavithai Vadevoo, S.M.; Baek, M.C.; Lee, B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials 2021, 278, 121137.


