Osteosarcoma (OS) is an aggressive bone tumor that mainly affects children and adolescents. OS has a strong tendency to relapse and metastasize, resulting in poor prognosis and survival. The high heterogeneity and genetic complexity of OS make it challenging to identify new therapeutic targets. Mesenchymal stem cells (MSCs) are multipotent stem cells that can differentiate into adipocytes, osteoblasts, or chondroblasts. OS is thought to originate at some stage in the differentiation process of MSC to pre-osteoblast or from osteoblast precursors. MSCs contribute to OS progression by interacting with tumor cells via paracrine signaling and affect tumor cell proliferation, invasion, angiogenesis, immune response, and metastasis. Extracellular vesicles (EVs), secreted by OS cells and MSCs in the tumor microenvironment, are crucial mediators of intercellular communication, driving OS progression by transferring miRNAs/RNA and proteins to other cells. MSC-derived EVs have both pro-tumor and anti-tumor effects on OS progression. MSC-EVs can be also engineered to deliver anti-tumor cargo to the tumor site, which offers potential applications in MSC-EV-based OS treatment.
1. Introduction
Osteosarcoma (OS) is the most predominant primary bone cancer, commonly occurring in the long bones of children and adolescents
[1]. OS is highly malignant and the major complications in OS arise due to a lack of immune response, leading to irregular bone growth and distant metastases, seen commonly in the lungs and liver. The current treatment approaches for OS are preoperative chemotherapy, surgical resection, and postoperative chemotherapy, which are effective in patients with localized OS. Conversely, patients with advanced, metastatic, and recurrent OS develop resistance to chemotherapy, which makes it difficult to treat, resulting in a poor prognosis
[2]. Despite multidisciplinary treatments, there has been no change in the prognosis during the past two decades. The overall 5-year survival rate of OS patients is 65% in the case of localized disease, while it is 20% in those with metastasis, and significantly lower in those with lung metastasis
[3]. The high heterogeneity and genetic complexity of OS make it challenging to identify new therapeutic targets
[4].
A thorough understanding of the tumor microenvironment (TME), especially the bone microenvironment (BME), cellular crosstalk, and the molecular mechanisms underlying tumor progression, is essential for drug design and for developing new drug molecules for OS treatment. The BME is composed of the extracellular matrix (ECM) and a variety of cells, which includes mesenchymal stem cells (MSCs), endothelial cells, macrophages, stem cells, fibroblasts, osteoblasts, osteoclasts, and osteocytes that are organized to maintain the bone rigidity and the structural as well as functional integrity of the bone niche. All these cells together play a crucial role in normal bone development and bone physiology and can also lead to osteosarcoma in aberrant conditions.
MSCs are multipotent, non-hematopoietic cells that have the potential to self-rejuvenate and to differentiate into different cell types, including muscle cells, hepatocytes, osteoblasts, adipocytes, chondrocytes, and stromal cells
[5][6][5,6]. MSCs interact with cancer and other cells in the tumor microenvironment via paracrine factors and through extracellular vesicles (EVs) to support tumor growth, progression, and metastasis. Contrary to their tumor-supportive role, they have also been implicated in tumor suppression. Hence, it is vital to understand the interaction of MSCs and OS cells in the tumor microenvironment to develop new and more effective OS therapy, and to overcome drug resistance. In this review, we discuss the role of MSCs in OS and highlight the reciprocal interaction of MSCs and OS cells in modulating each other’s functions, with a focus on the role of extracellular vesicles in MSC–OS communication and their therapeutic applications.
2. Mesenchymal Stem Cells’ Role in Osteosarcoma
MSCs are an important source of adult stem cells, present in many tissues, especially in bone marrow, and adipose tissue, and are involved in tissue repair and healing. MSCs play a significant role in OS pathogenesis, from the possible cells of origin of OS to its supportive role in OS growth, progression, metastasis, and drug resistance. OS is assumed to originate at some stage in the differentiation process of MSC to pre-osteoblast and the stage of differentiation of MSC affects the OS phenotype
[7][8][7,8].
2.1. Cellular Origin of OS
A better understanding of the cellular origin of OS is essential to improve the outcomes of patients. There are controversial reports about the cellular origins of OS. Some propose that OS develops from MSC; by contrast, others suggest osteoblast precursor cells as the cells of origin of OS
[9].
MSCs can differentiate into osteoblasts, chondroblasts, or fibroblasts, and OS can have an osteoblastic, chondroblastic, or fibroblastic phenotype, based on the predominant cancer cell type, which suggests that MSC might be the cells of origin of OS
[10][11][10,11]. There is evidence suggesting that OS originates at some point during the differentiation of MSC to pre-osteoblast
[7][8][7,8]. The argument that MSC is a cell of origin of OS is supported by studies in murine models, where MSCs have been shown to transform into OS by genetic loss of cdkn2 locus
[12]. A similar transformation of human MSCs into OS has been seen in the simultaneous knockdown of Rb and overexpression of c-Myc
[13].
Although in vitro and in vivo genetic models support the proposition that the unusual differentiation of pre-osteoblasts may be the origin of OS, recent studies suggest that osteogenic progenitors rather than undifferentiated MSCs represent the OS cells of origin
[8][14][8,14]. The transformation of human osteo-progenitor cells into OS is possible due to the overexpression of the MET gene
[15]. Other studies have shown that the osteogenic differentiation stage of bone marrow MSCs (BM-MSCs) inflicts the phenotype of in vivo sarcoma development, implying that BM-MSC-derived osteogenic progenitors might be the cells of origin for OS
[16]. Several in vivo genetic model studies support the idea that the abnormal differentiation of osteoblasts is the origin of OS cells. The debate arises from the notion that under physiological conditions, it is challenging to discriminate between MSCs and osteoblasts. While MSCs have the potential to differentiate into osteoblasts, they can transform into OS cells in a specific bone microenvironment
[17].
To date, there is no conclusive evidence as to whether OS arises because of the transformation of MSCs, or osteoblastic lineage-committed cells (pre-osteoblasts). The use of advanced gene modification technology and improved gene-editing tools at the cell lineage level may help to explain the origin of OS. Understanding the cellular origin of tumors is key to improving preclinical models for testing and studying their behavior and treatment approaches, which can accelerate translational research efforts to advance outcomes for patients with OS.
2.2. MSCs in OS Microenvironment
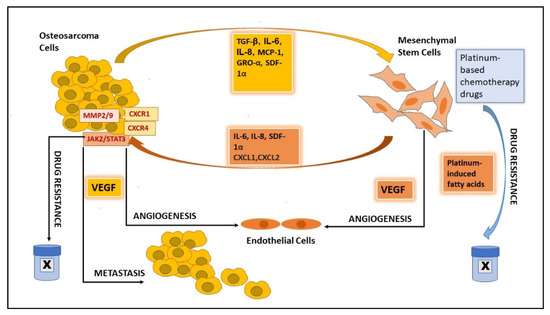
OS grows in a bone microenvironment. This is a very specialized, complex, and highly dynamic environment composed of bone cells (osteoclasts, osteoblasts, osteocytes), stromal cells (MSCs, fibroblasts), vascular cells (endothelial cells and pericytes), immune cells (macrophages, lymphocytes), and a mineralized ECM. Crosstalk between OS, bone microenvironment, and ECM involves several signals, induced by multiple cytokines, chemokines, and growth factors carried by EVs. The tumor microenvironment contributes significantly to the development of OS, and MSCs in the TME play an important role in OS growth, progression, metastasis, and drug resistance
[18]. Two types of MSCs, from normal tissue and tumor tissue, can accelerate OS growth
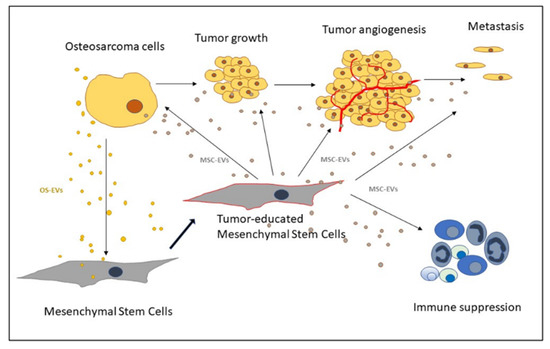
[19]. Normal-tissue MSCs are recruited to the TME and educated to undergo heterogeneous differentiation into a pro-tumor phenotype (
Figure 1). Despite having similar morphological features and differentiation abilities, tumor MSCs derived from OS tissue are more potent effectors of tumor progression compared to normal tissue MSCs.
Figure 1. Schematic representation of extracellular vesicles (EVs)-mediated communication between mesenchymal stem cells and osteosarcoma cells (OS) in the OS microenvironment.
2.3. Recruitment of MSCs to the Tumor Site
OS cells communicate with their microenvironment via paracrine signals that induce the homing of MSCs to the tumor site, where it undergoes changes and produces inflammatory signals that affect the tumor growth, the angiogenesis process, and the immune response (
Figure 1). Transforming growth factor (TGF-β) and stromal-derived factor (SDF1) are reported to induce the migration of MSCs towards OS cells, while MCP-1, GRO-α, and TGF-β, in addition to effecting the chemotaxis of MSC, also induce the differentiation of MSCs into cancer-associated fibroblasts, resulting in mesenchymal-to-amoeboid transition
[20].
Besides growth factors, proteases derived from ECM and chemokines play a vital role in the recruitment of MSCs at the tumor site. The activation of plasminogen, through the coupling of the Urokinase plasminogen activator and its receptor, induces the migration of MSCs to the tumor site. Moreover, higher plasminogen activity in solid tumors is associated with a greater migration of MSCs, indicating that MSCs recruitment is dependent on plasminogen activity. However, the underlying mechanism through which MSC recruitment is enhanced through plasminogen activation has not been fully explored
[21]. MMP-1 is another ECM protease that facilitates MSC recruitment to tumor site
[22].
2.4. OS Microenvironment-Induced Changes in MSCs
MSCs that migrate to the tumor site or are present in TME change to acquire a phenotype that promotes tumor progression and metastasis (
Table 1)
[23]. We have shown that OS-derived EVs bring about epigenetic changes and an increased expression of
VEGF-A in MSCs
[24]. OS cells cause the upregulation of IL-6 and VEGF in MSCs, thereby maintaining their stemness, which supports tumor growth and migration
[25][26][25,26]. Hypoxic conditions in the tumor microenvironment induce tumor cells to switch to anaerobic glycolysis, which results in ECM acidification, which in turn helps in the conversion of normal MSCs into tumor MSCs. Avnet et al. showed that short-term acidosis-exposed MSCs induce NF-κB pathway activation, clonogenicity, and invasion in OS cells in co-culture
[27]. Moreover, acidosis was shown to induce a tissue remodeling phenotype of MSC, with an increased expression of colony-promoting factors, chemokines (CCL5, CXCL5, and CXCL1), cytokines (IL6 and IL8), and CXCR4. They further reported that the increased expression of IL6 and IL8 was seen only in normal stromal cells, but not in OS cells, which was similarly confirmed in tumor-associated stromal cells
[27]. The modified MSC phenotypes can also account for the development of chemoresistance via IL6 secretion, and this mechanism holds potential for future therapeutic interventions aimed at targeting OS
[27].
Table 1. Interaction of mesenchymal stem cells (MSCs) and osteosarcoma cells (OS).
| OS Signal |
Effect on MSCs |
MSC Signal |
Effect on OS |
Reference |
52][66,67]. The detection of tumor-specific EVs in blood circulation may serve as a diagnostic tumor marker
[48][63].
The development of OS and tumor driving specific genetic alterations are incompletely understood so far. The alterations at the epigenetic level could be the early event occurring in the transformation of MSCs during OS development. Recently, we presented the EV-mediated intercellular crosstalk between MSC and OS, demonstrating that OS-EVs modulate the epigenetic status of MSC, through the hypomethylation of long interspersed nuclear element 1 (
LINE1), whereas an opposite effect was seen in pre-osteoblasts. Our results indicate that MSCs, but not pre-osteoblasts, are susceptible to OS-EV-mediated epigenetic transformation. Furthermore, OS-derived EVs influenced the AD-MSC’s expression of matrix metallopeptidase 1 (
MMP1), vascular endothelial growth factor A (
VEGF-A), and intercellular adhesion molecule 1, which are related to bone microenvironment remodeling
[24].
3.1. OS-EVs
OS cell-derived exosomes affect the tumor microenvironment by carrying microRNAs, which induce bone remodeling and tumor angiogenesis by promoting osteoclasts differentiation and bone resorption activity. The different proteins, microRNAs, and other non-coding RNAs detected in the EVs cargo derived from OS cells and MSCs that are involved in OS pathogenesis are summarized in
Table 2.
Table 2. Role of microRNA, non-coding RNA, and proteins associated with osteosarcoma (OS) and mesenchymal stem cell (MSC)-derived extracellular vesicles (EVs) in OS pathogenesis.
| Source of EVs |
miRNAs/other RNAs in EVs |
Proteins in EVs |
Function |
Reference |
| |
|
Exosomes |
Increase in COLGALT2 and proliferation. |
Wang et al. 2020 [28] |
| OS cells |
miR-146a-5p, miR-10b-5p, miR-143-3p, miR-382-5p, miR-150-5p, miR-125b-5p, miR-27a-3p, miR-145-5p, miR-26a-5p, miR-93-5p, miR-21-5p, miR-92a-3p, and miR-106a-5p |
serpin-E1, serpin-F1, TIMP-1, thrombospondin-1, urokinase-type plasminogen activator (uPA), VEGF, pentraxin-3, PDGF-AA, angiopoietin-2, coagulation factor-III, CD26, CD105, endostatin, endothelin-1, and HB-EGF |
Angiogenesis. |
Perut et al. 2019 [53][68] |
| |
|
CM 1/co-culture |
Increase in MMP2/9; STAT3 activation. Increased proliferation, invasion, and metastasis. |
Wang et al. 2017 [29] |
| OS cells & tissue |
lncRNA OIP5-AS1 |
|
Angiogenesis. |
Li et al. 2021 [54][69] |
IL-8
(co-culture) |
Increased IL-8. |
IL-8
(co-culture) |
Increased IL-8.
Increased metastatic potential. |
Kawano et al. 2018 [30] |
| OS cells |
miR-148a-3p and miR-21-5p |
|
TME remodeling. |
Raimondi et al. 2020 [49][64] |
|
|
IL-6
CM/co-culture |
Increase in MMP2/9; JAK2/STAT3 activation. Increased proliferation, migration, and doxorubicin resistance. |
Lu et al. 2021 [31] |
| OS cells |
|
TGFβ |
Increase IL6 in AD-MSCs, tumor growth, STAT3 activation, and lung metastasis.
Autophagy. |
Baglio et al. 2017, Tu et al. 2012, Huang et al. 2020 [23][33][55][23,33,70] |
MCP-1, GRO-α, and TGFβ |
Mesenchymal-to-amoeboid transition.
Increase in MCP-1, GRO-α, IL-6, and IL-8 in the tumor environment. |
|
| BM-MSC |
lncRNA MALAT1 | Increased migration, invasion, and trans-endothelial migration. |
| Pietrovito
|
Proliferation, invasion, and migration of OS cells via lncRNA MALAT1/miR-143/NRSN2/Wnt/β-Catenin Axis.et al. 2018 [20] |
| Li et al. 2021 | [ | 56 | ] | [71] |
OS-EVs |
LINE-1 hypomethylation increased VEGF-A. |
|
|
Mannerström et al. 2019 [24] |
| |
|
CM |
| BM-MSC |
non-coding RNA PVT1 |
|
OS migration by upregulating ERG and sponging miR-183-5p in OS cells. |
Zhao et al. 2019 [57][72] |
STAT3 activation.
Promote survival and drug resistance. |
Tu et al. 2016 [25] |
| BM-MSC |
miR-206 |
|
Tumor suppression and apoptosis. |
Zhang et al. 2020 [58][55] |
|
|
MSC-EVs
(under stress) |
| BM-MSC | Increased migration. Apoptosis resistance. |
microRNA-208a
LCP1 | Vallabhaneni et al. 2016 |
|
OS cell migration & invasion.
OS proliferation and metastasis via the JAK2/STAT3 pathway.[32] |
| Co-culture |
Increased TGFβ. |
Co-culture
IL-6 |
Increased OS proliferation, stemness & migration. |
Cortini et al. 2016 [26] |
| |
|
IL-6 |
STAT3 activation. Increased proliferation and metastasis. |
Tu et al. 2012 [33] |
| CM/TGFβ |
Increased IL-6, VEGF.
Inhibit osteogenic differentiation. |
|
|
Tu et al. 2014 [34] |
| |
|
IL-8 |
CXCR1/Akt activation.
Promotes metastasis. |
Du et al. 2018 [35] |
| EVs/TGFβ |
Increased IL-6. |
Tumor-educated MSC |
Activation of STAT3 signaling. |
Baglio et al. 2017 [23] |
| |
|
EVs |
Cell growth under hypoxia. Activation of PI3K/AKT & HIF-1α. |
Lin et al. 2019 [36] |
| |
|
EVs |
Activation of Hedgehog signaling. Tumor growth. |
Qi et al. 2017 [37] |