Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Georges Nemer | + 3715 word(s) | 3715 | 2021-10-09 04:42:38 | | | |
| 2 | Georges Nemer | Meta information modification | 3715 | 2021-10-10 13:09:51 | | | | |
| 3 | Jessie Wu | Meta information modification | 3715 | 2021-10-11 03:12:05 | | | | |
| 4 | Jessie Wu | Meta information modification | 3715 | 2021-10-11 03:27:58 | | | | |
| 5 | Jessie Wu | Meta information modification | 3715 | 2021-10-11 05:36:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nemer, G. Pharmacogenomics Variability of Lipid-Lowering Therapies. Encyclopedia. Available online: https://encyclopedia.pub/entry/14927 (accessed on 07 February 2026).
Nemer G. Pharmacogenomics Variability of Lipid-Lowering Therapies. Encyclopedia. Available at: https://encyclopedia.pub/entry/14927. Accessed February 07, 2026.
Nemer, Georges. "Pharmacogenomics Variability of Lipid-Lowering Therapies" Encyclopedia, https://encyclopedia.pub/entry/14927 (accessed February 07, 2026).
Nemer, G. (2021, October 10). Pharmacogenomics Variability of Lipid-Lowering Therapies. In Encyclopedia. https://encyclopedia.pub/entry/14927
Nemer, Georges. "Pharmacogenomics Variability of Lipid-Lowering Therapies." Encyclopedia. Web. 10 October, 2021.
Copy Citation
The exponential expansion of genomic data coupled with the lack of appropriate clinical categorization of the variants is posing a major challenge to conventional medications for many common and rare diseases. To narrow this gap and achieve the goals of personalized medicine, a collaborative effort should be made to characterize the genomic variants functionally and clinically with a massive global genomic sequencing of “healthy” subjects from several ethnicities. Familial-based clustered diseases with homogenous genetic backgrounds are amongst the most beneficial tools to help address this challenge
familial hypercholesterolemia
pharmacogenomics
1. Introduction
Familial hypercholesterolemia (FH) is the most common and first acquired pathology of lipoprotein metabolism to be characterized genetically and clinically [1]. It is identified by elevated levels of absolute low-density lipoprotein cholesterol (LDL-C) in the blood, early-onset of atherosclerotic cardiovascular diseases (ASCVD), fat accumulation in external tissues, and tendon and cutaneous xanthomas [2]. Globally, heterozygous FH afflicts around one in 250–500 individuals, with a more noticeable predominance in special communities, including the Christian Lebanese, French-Canadian, Finnish, and Afrikaner [3]. Medical symptoms of the severe phenotype, a homozygous FH, initiates at the early stages of childhood with a predicted incidence of one in a million. The intensity of FH complications such as coronary ostium and aortic root predominantly rely on total LDL-C levels [4][5]. Clinical examination of FH can be verified based on premature cardiovascular diseases (CVD), physical marks, and history of raised cholesterol levels. In addition to the serum lipids analysis, various systemic diagnostic guidelines suggest cascade genomic examining to detect FH and confirm the polymorphisms in family members up to the third degree, including the Dutch Lipid Clinic Network—Make Early Diagnosis to Prevent Early Deaths (Dutch-MEDPED) [6]. Genetic testing could facilitate early recognition and treatment of undiagnosed, untreated FH patients and is known to provide a better prognosis of the disease.
Inherited disease-causative impairments of the low-density lipoprotein-receptor gene, (LDLR) present in 70–90% of subjects, and, less commonly, the apolipoprotein B gene (APOB), as well as proprotein convertase subtilin/Kexin, member nine genes (PCSK9) have been linked to raised lipoprotein cholesterol in FH (190–400 mg/dL) [3]. Additional genes encoding the LDLR-adaptor protein 1 (LDLRAP1) and apolipoprotein E (APOE) can infrequently correlate with cholesterol homeostasis and promote the development of autosomal recessive FH [4]. APOB and APOE genes are responsible for encoding ApoB-100 and ApoB-48 isoforms as well as ApoE, respectively, which are the elemental apolipoproteins of the LDL-C and are the protein ligands of LDLR. PCSK9 gene encodes member 9 of the PCSK family that involves the lysosomal degeneration and coordination of LDLR. The LDLRAP1 protein encoded by the LDLRAP1 gene has a phosphotyrosine binding domain that interacts and harmonizes the LDLR activity. The physiological uptake and catabolism of fats are essentially mediated by hepatic LDLR, which is encoded via the LDLR gene [3][4]. Interestingly, the number of variations in LDLR and associated genes related to the clinical manifestations of FH is uniformly rising.
For a long time, there was an apparent focus on investigating LDLR variants to recognize the impact on the medical, biochemical, and pathological phenotypes of FH monogenic dysfunctions. It is noteworthy that the significant phenotypic diversity of lipids and coronary artery disorders depends on the nature of FH genetic defects. These defects are modulated, however, by various genetic and epigenetic factors, and, thus, various pathological genotypes can differentially impact the circulating levels of LDL-C [7][8]. For instance, a nonsense variant in the LDLR (c.2043 C>A, p. cys681X) was predominantly combined with familial hypercholesterolemia in nearly 82% of Lebanese cases. This Lebanese allele leads to a LDLR loss-of-function (null) defect and attenuates hepatic metabolism and removal of LDL-C and is believed to lead to a very severe phenotype [9]. Paradoxically, the mutation is a founder mutation in the Lebanese population and was encountered in Lebanese individuals with normal cholesterol levels. This indicates the presence of unrecognized variants and/or an epigenetic signature that counters the effects of the deleterious LDLR mutation in these cases [10]. Consequently, genetic diagnostic screening of disease-causative mutations, considered the gold standard for FH detection, is not enough but should be coupled with whole-genome sequencing and/or methylation analysis to further stratify affected members within familial cases.
Despite the prevalence of FH and the significance of early determination and management of the condition, only 15–20% of FH subjects are diagnosed by medical examination. Untreated patients with heterozygous FH have a nearly 20-fold higher raised incidence of premature coronary artery disease relative to cases without FH [11]. Coronary artery disease and heart attacks restrict coronary blood flow, causing the pumping chamber to enlarge, widen, and attenuate. Ultimately, this damage will lead to ischemic cardiomyopathy, potentially reducing the ability of cardiomyocytes to pump blood [12]. The earliest clinical mark of the disease regularly happens throughout the third decade of growth, particularly in severe cases with LDLR-negative mutational status, such as fatal myocardial infarction [5]. Appropriate identification and management could control the lipid levels and limit the life-threatening complications of FH. A current examination documented that early introduction of lipid-lowering medicine throughout childhood and adolescents in cases with FH can ameliorate the pathological progression and reduce the incidence of ASCVD, explaining the considerable advantage of immediate FH treatment [13]. Numerous studies have revealed that only 10% reached the recommended cholesterol levels even though most patients receive the maximum tolerated cholesterol-modulating drugs [14].
The pharmacological variation among FH patients has been linked to the genomic single nucleotide polymorphisms (SNPs) of genes associated with cholesterol catabolism and biosynthesis [15]. In extreme cases of FH, genetic screening could potentially be used to examine the response variability and, therefore, to effectively personalize the therapeutic care plan for the anti-lipid interventions and CVD risk preventions. Under this scenario, this review will discuss the management of familial hypercholesterolemia with the standard and innovative therapeutic strategies from the prospect of pharmacogenomics and its link to the causative genetic mutations underlying the phenotype.
2. FH Management
The main goal of FH therapy is to reduce relative LDL-C by more than 50% or to lower LDL-C to 100 mg/dL in adults without ASCVD. For FH subjects with ASCVD or major CVD risk, the 2019 ESC/EAS guidelines recommend a more than 50% reduction of LDL-C or less than 55 mg/dL of absolute LDL-C [6]. Therapeutic lifestyle modifications such as restricted diet, regular physical training, limiting alcohol intake, and smoking cessation are all fundamental in the controlling of FH [16]. In addition, patients should be counseled to maintain healthy blood sugar, blood pressure, and weight. Despite the paramount importance of non-pharmacological management in all FH patients, optimizing cholesterol levels and preventing CVD are hardly achieved without pharmacological interventions [5].
Currently, β-hydroxy-β-methylglutaryl coenzyme A reductase (HMGCR) inhibitors at the highest tolerable dose are strongly recommended to be initiated immediately at diagnosis in all FH adults. Monotherapy, daily doses of the aggressive statins, and HMGCR inhibitors, including atorvastatin 40–80 mg and rosuvastatin 20–40 mg orally per day, are expected to decrease LDL-C approximately 50–60%, as reported in various cholesterol-lowering studies [11][14]. When the target fails to be achieved, stepwise intensification of anti-lipid medications should be considered. Ezetimibe, a cholesterol uptake blocker, can decrease the LDL-C by nearly 25% and is recommended as an adjunctive second-line treatment [6][11].
PCSK9-based medications are a great breakthrough in FH pharmacotherapy, with a reduction in LDL-C ranging from 25% to 30%. The anti-PCSK9 monoclonal antibodies, evolocumab, and alirocumab, should be initiated if maximum intense statins and ezetimibe fail to sufficiently control the lipid profile in FH cases with a major risk of cardiomyopathies [6]. In 2013, the Food and Drug Administration (FDA) approved using a microsomal triglyceride transfer (MTP) inhibitor, lomitapide, and an oligonucleotide agent, mipomersen, as add-on medications to the classic anti-lipids in adults with the severe FH phenotype. Lipoprotein apheresis should be offered for individuals with either homozygous FH or severe-uncontrolled heterozygous FH, which may reduce 60–80% of LDL-C [6]. The cholesterol metabolism pathway and pharmacological targets for classical and novel lipid-lowering therapies are schematically summarized in Figure 1.
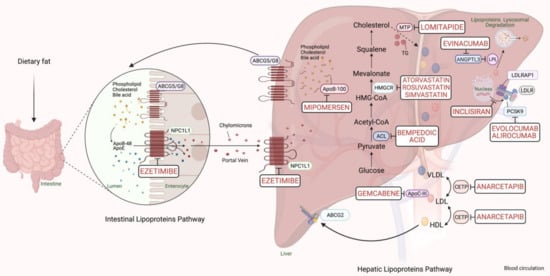
Figure 1. Scheme representing the cholesterol metabolism pathway and pharmacological targets for classical and novel lipid-lowering therapies (generated with BioRender.com). Unique hepatic and intestinal transporters (ABCG5 and ABCG8) release bile acids, phospholipids, and cholesterol into the biliary system. Inversely, the NPC1L1 protein modulates cholesterol returning to hepatocytes. Ezetimibe inhibits cholesterol entry into the intestine and liver by the NPC1L1 transporter. Next, chylomicrons are generated through the assembly of TG, cholesterol, and ApoB-48, and released into the blood circulation (see intestinal lipoproteins pathway). The speed-limiting enzyme of endogenous cholesterol synthesis, HMGCR (see hepatic lipoproteins pathway), is inhibited via statins. The activated bempedoic acid decreases the hepatic synthesis of acetyl CoA and cholesterol catabolism by blocking the ACL protein. ApoB-100, phospholipid, ApoC-III, and cholesterol assembly into VLDL depend on the activity of MTP, which is blocked by lomitapide. The degradation of hepatic messenger ribonucleic acid (mRNA) transcript of ApoB-100 and ApoC-III is mediated by mipomersen and gemcabene, respectively. The heteroexchange of TGs and cholesteryl esters between ApoB-lipoproteins particles relies on CETP activity, which is blocked by CETP inhibitors such as anacetrapib. LDLR interacts and removes LDL-cholesterol from the blood circulation with the assistance of LDLRAP1. Lysosomal catabolism of LDLR is mediated by PCSK9. Anti-PCSK9 antibodies, including evolocumab, alirocumab, and inclisiran inhibit the endogenous production and release of PCSK9. Evinacumab blocks the inhibition of hepatic lipoprotein lipase activity by ANGPTL3. Abbreviations: ApoB-48/100, Apolipoprotein B protein member 48 & 100; ApoC-III, Apolipoprotein C protein, member III; ApoE, Apolipoprotein E protein; HDL, High-density lipoprotein cholesterol; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein; TG, triglyceride; LDLR, LDL-receptor; LDLRAP1, LDLR-adaptor protein, member 1; ABCG2, atp-binding cassette, subfamily g, member 2; HMGCR, β-hydroxy-β-methylglutaryl Coenzyme A Reductase; NPC1L1, Niemann-Pick C1-like 1 transporter protein; ACL, adenosine triphosphate citrate lyase; MTP, microsomal triglyceride transfer protein; LPL, lipoprotein lipase; CETP, cholesteryl ester transfer protein; ANGPTL3, angiopoietin-like protein 3; PCSK9, proprotein convertase subtilin/kexin 9 protein.
For familial genotyped cases, the current therapeutic protocols suggest maintaining LDL-C below 135 mg/dL for children aged 10 or older and reducing LDL-C by 50% for younger ages. A healthy lifestyle could achieve this goal in conjugation with a mild statin regimen, subject to the dosage increase, to reach the targeted cholesterol levels [6][16]. Nevertheless, various FH cases are still not controlled despite aggressive therapeutic interventions. Contemporary information from the International Pediatric FH Register revealed that 23% of heterozygous FH children on statins had not reached the targeted LDL-C goal (below 135 mg/dL) [17].
The wide variation of cholesterol-lowering therapies in terms of the potential adverse effect and therapeutic response is one of the main challenges in clinical practice, especially in FH patients. In addition to clinical and environmental factors, including race, gender, age, smoking, and adverse consequences, genomic phenotypes of LDLR, APOB, and PCSK9 can potentially modulate the sensitivity of anti-lipids. Over the previous decade, many pharmacogenomics and genome-wide association studies (GWASs) have recognized numerous genetic variations that can affect the therapeutic potency (anti-lipid pharmacodynamics), drug absorption, metabolism, excretion (anti-lipid pharmacokinetics), and anti-lipid toxicity pathways [3][18]. Accordingly, therapeutic efficiency and safety, and patient quality of life could be promoted through personalized genomic examination, which is designed to predict the therapeutic response of FH management.
3. Pharmacogenomics of Statin in FH
The primary and secondary prevention of CVD and the cornerstone medication in patients with FH are via HMGCR inhibitors [5][6]. Statins could potentially decrease the plasma levels of atherosclerotic LDL-C via competitively inhibiting the HMGCR (Figure 1) [11]. The inhibition of this protein reduces the hepatic synthesis of cholesterol and, thereby, enhances LDLR production. Subsequently, the elevated expression of LDLR on the hepatocytic membrane will increase the cellular uptake of cholesterol from the bloodstream, mainly by the liver. Furthermore, the secretion of ApoB-containing lipoproteins, LDL, and very-low-density lipoprotein (VLDL), as well as triglycerides from hepatocytes, may also be lowered via statins [11]. The lifelong overburden of high cholesterol makes patients with FH highly susceptible to the risk of CVD and significantly reduces their life expectancy [2]. Although statins robustly diminish cholesterol in addition to CVD morbidity and mortality by 20–30% in normal individuals, their efficacy is predominantly weaker in FH subjects [5]. Genetic variations combined with non-adherence due to statin myotoxicity or hepatotoxicity may cause pharmacological variability among patients. We will divide the variants according to the effect they have on either the pharmacodynamics or the pharmacokinetics of these drugs.
3.1. SNPs Linked to Pharmacodynamics of Statins in FH
The hepatocyte endocytosis of lipoproteins is mediated mainly by LDLR in addition to other processing associated proteins, including PCSK9, APOE, and LDLRAP1. SNPs in the LDLR could selectively reshape the anti-lipids therapeutic outcome and the incidence of FH and coronary artery conditions. Therefore, the pharmacogenetic analysis principally concentrates on discovering these mutations, as reviewed in Table 1 [19][20][21][22][23][24]. Polisecki and colleagues observed a strong association between the serum-baseline cholesterols and statin efficacy in terms of coronary artery disease risk in FH patients carrying an LDLR polymorphism (rs1433099, c.44857C>T) [25]. The 3′-untranslated region (3′-UTR) of LDLR has been found to play a basic role in the anti-lipids mediated-LDL-C reduction through stabilizing the LDLR mRNA. Polymorphisms at the 3-UTR loci have been linked to lipid baselines, LDLR activity, and CVD [26]. Interestingly, subjects with mixed LDLR and HMGCR haplotypes have more prominent attenuations in optimizing desired cholesterols than those carrying a single LDLR mutation [27]. The cholesterol-lowering potency of pravastatin has also been modulated by another LDLR genetic defect (rs5925, c.2052T>C) [28]. Recently, LDLR stop-gained pathogenic variants (c.2027delG, p. Gly676Alafs*33) have been discovered to be correlated with anti-lipid efficacies including statins, ezetimibe, and clinical manifestations of FH [29].
Several investigations have noticed that the physiological effect of lipid-modifying drugs, especially statin, and the lipid profile in heterozygous FH subjects is affected by the presence of LDLR variants and the type of mutation. Individuals carrying a null mutation (deletions result in a frameshift and premature stop codon) were observed to have diminished LDL-C responses with elevated cholesterols compared to patients carrying a defective (non-frameshift small insertions or deletions) or without mutation [30][31][32][33][34].
Table 1. Pharmacogenomics variations associated with statin response in familial hypercholesterolemia patients.
| Gene | Significant Mutation * | Patients | Population | Sample Size | Treatment and Daily Dose | Clinical Findings | Author, Year (References) |
|---|---|---|---|---|---|---|---|
| LDLR | FH1 (C206G) & FH2 (G408A) |
Het-FH | Afrikaners | 20 | Simvastatin 40 mg | TC reduction is higher in patients with FH2 than FH1 | Jeenah et al., 1993 [19] |
| LDLR | C660X, D147H, & 652delGGT | Het-FH | Israeli | 64 | Fluvastatin 40 mg | Reduction of LDL-C, apoA, and elevation of HDL-C depend variously on LDLR mutations | Leitersdorf et al., 1993 [23] |
| APOE | E2,3, & 4 alleles | Het-FH | Canadian | 49 | Lovastatin 80 mg | Statin sensitive is higher in men with E4 than E3 or E2 or women with any APOE phenotype | Carmena et al., 1993 [35] |
| LDLR | FHTONAMI-1 (Del exon15) &FHKANAZAWA (C665T) | Het-FH | Japanese | 12 | Pravastatin & cholestyramine | LDL-C reduction is higher in patients with FHKANAZAWA than FH1 FHTONAMI-1 | Kajinami et al., 1998 [20] |
| LDLR | W66G, C646Y, & deletion>15 kb | Het-FH | Canadian | 63 | Simvastatin 20 mg | LDL-C reduction is higher in patients with C646Y & deletion > 15 kb than W66G | Couture et al., 1998 [21] |
| LDLR | Severe and mild LDLR | Het-FH | British | 42 | Simvastatin + bile acid sequestrant | LDL-C is higher in patients with severe than mild mutation | Sun et al., (1998) [33] |
| LDLR | Null and defective LDLR | Het-FH | British | 109 | Simvastatin | LDL-C reduction is higher in patients with defective than null mutation | Heath et al., (1999) [31] |
| LDLR | AvaII (rs5925T>C), HincII (rs688C>T), & PvuII (rs2569542A>G) | Het-FH | Brazilian | 55 | Fluvastatin 40–80 mg | LDL-C, TC, & ApoB reduction is higher in patients with AvaII & PvuII than HincII | Salazar et al., 2000 [22] |
| LDLR | Null and defective LDLR | FH | Spanish | 55 | Simvastatin 20 mg | Low HDL-C & poor statin response are higher in patients with defective than null mutations | Chaves et al., (2001) [32] |
| APOE | E4 allele | Het-FH | British | 19 | Atorvastatin 10 mg + bile acid sequestrant | Poor statins response is high in patients with E4 phenotype | O’Neill et al., 2001 [36] |
| LDLR | Null and defective LDLR | Het-FH | Canadian | 63 | Atorvastatin 20 mg | LDL-C reduction is higher in patients with null than defective mutation | Vohl et al., (2002) [37] |
| LDLR | G1775A, G1646A, & C858A |
Het-FH | Greek | 49 | Atorvastatin 20 mg | LDL-C & ApoB reduction is higher in patients with G1775A than G1646A & C858A | Miltiadous et al., 2005 [24] |
| MTP | c.493 GT | Het-FH | Spanish | 222 | Atorvastatin 20 mg | High reduction of TG in men and low reduction of VLDL & TG in women with c.493 GT allele | García-Garc ía et al., 2005 [38] |
| CETP | −867 and Ex14/I405V | Het-FH | Israeli | 76 | Fluvastatin 40 mg | LDL-C reduction is high among CETP & MDR1 mutants | Bercovich et al., 2006 [39] |
| MDR1 | c.(G2677T) and c.(C3435T) | ||||||
| LDLR | Null and defective LDLR | Het-FH | Spanish | 811 | Simvastatin or atorvastatin 80 mg ± bile acid sequestrant | PCVD & TC is higher in patients with null than defective mutations | Alonso et al., 2008 [40] |
| ABCG2 | rs2231142 | FH | Chinese | 386 | Rosuvastatin 10 mg | High LDL-C reduction among patients with AA genotype | Hu et al., 2010 [41] |
| LDLR | Null and defective LDLR | FH | Spanish | 387 | Maximum statin doses ** + ezetimibe 10 mg | Poor LLT response & high PCVD in patients with null than defective mutations | Mata et al. (2011) [42] |
| LDLR | W556R | Twins with Hom-FH and parents with Het-FH (one family) | Turkish | 4 | Simvastatin 40 mg + ezetimibe 10 mg or LDL apheresi | Hom-FH have a low LDL-C reduction and high statin resistance, but Het-FH respond to statin with 60% LDL-C reduction | Schaefer et al., 2012 [43] |
| CYP3A4 | rs2740574 | FH | Chilean | 142 | Atorvastatin 10 mg | High statin sensitivity among patients with CYP3A4 mutations | Rosales et al., 2012 [44] |
| ANRIL | rs1333049 | FH with CVD | Pakistani | 611 | Atorvastatin 10, 20 or 40 mg | High LDL-C, TC, & TG reduction in patients with CC genotype | Ahmed et al., 2013 [45] |
| LDLR | Null (W66G) and defective (C646Y) LDLR | Het-FH | Brazilian | 156 | Atorvastatin 10, 20 or 40 mg | LDL-C reduction is more in patients with defective than with null mutation | Santos et al., 2014 [30] |
| POR | rs1057868 | FH | Greek | 105 | Atorvastatin 10, 20 and 40 mg | High LDL-C & TC reduction in patients with 1/1 genotype | Drogari et al., 2014 [46] |
| MYLIP | rs9370867 | Het-FH | Brazilian | 156 | Atorvastatin 10–80 mg ± ezetimibe 10 mg | High LDL-C reduction in patients with AA genotype | Santos et al., 2014 [47] |
| PSCK9 | E32K | Hom-FH | Japanese | 1055 | Atorvastatin 80 mg & ezetimibe 10 mg | PSCK9 gain-of-function variants significantly worsen LDLR phenotype and decrease LDL-C reduction | Mabuchi, et al., 2014 [48] |
| LDLR | Double allele | ||||||
| LDLR | Null and defective LDLR | FH | Spanish | 4132 | Maximum statin doses ** + ezetimibe 10 mg | Poor LLT response & CVD events are higher in null than in defective mutation | Perez de Isla et al., 2016 [14] |
| LDLR | p.(Cys155Gly) | Hom-FH | Belgian | 8 | Atorvastatin 80 mg, ezetimibe 10 mg, cholestyramine | LLT efficacy is attenuated in patients with nonsense LDLR mutations | Sanna et al., 2016 [34] |
| HMGCR | rs3846662 | Het-FH | French Canadian | 106 | Statin + LLTΩ | Poor statin response among HMGCR mutants | Leduc et al., 2016 [49] |
| LDLR | W87G, C368Y, T726I, G2fsX214, D47N, N97H, E101K, C216fsX, L582P, C667Y, & LDLR-17-18 del | Het-FH | American and Canadian | 139 | Atorvastatin 40/80 mg, rosuvastatin 20/40 mg or simvastatin 40/80 mg, + Bococizumab 0.25, 1, 3, or 6 mg/kg |
Bococizumab effecacy is higher than statin in reducing LDL-C across LDLR & APOB variants | Fazio et al., 2018 [50] |
| APOB | R3527Q | ||||||
| LDLR | Het-LDLR mutation | FH | Spanish | 22 | Maximum statin doses ** ± ezetimibe 10 mg | LDL-C reduction is higher in patients with p.(Leu167del) mutation than LDLR | Bea et al., 2019 [51] |
| APOE | p.(Leu167del) | ||||||
| LDLR SLCO1B1 ABCB11 CYP3A5 |
rs28941776 c.(521T>C; SLCO1B1*5) & c.(388A>G; SLCO1B1*1B) rs2287622 CYP3A5*3 |
FH | Caucasian | 1 | Rosuvastatin 40 mg & ezetimibe 10 mg | Loss-of-function mutations enhance statin myotoxicity and delay its response | Dagli-Hernandez et al., 2021 [52] |
| LDLR | c.(2027delG), p. (Gly676Alafs*33) | FH (2 families) | Saudi | 12 | Statin + ezetimibe | Clinical manifestations and poor LLT response depend on LDLR variants | Awan et al., 2021 [29] |
* Borderline significance (p < 0.05). ** Maximum tolerated dose of statin: simvastatin 80 mg, pravastatin 40 mg, lovastatin 80 mg, fluvastatin 80 mg, atorvastatin 80 mg, or rosuvastatin 20–40 mg. Ω LLT including ezetimibe, fibrates, statins, lomitapide, PCSK9 inhibitors, bile acid sequestrants (e.g., cholestyramine and colestipol), or niacin. Abbreviations: LLT; lipid-lowering therapies; PCVD, premature cardiovascular diseases; FH, familial hypercholesterolemia; Het-FH, patients with heterozygous FH; Hom-FH, patients with homozygous FH; ApoB, Apolipoprotein B protein; HDL-C, High-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol; TG, triglyceride; LDLR, Low-density lipoprotein receptor; APOB, Apolipoprotein B; ABCG2, atp-binding cassette, subfamily g, member 2; MDR1, multidrug resistance mutation 1; CYP3A4, Cytochrome P450, family 3, subfamily A, member 4; ANRIL, antisense non-coding RNA in the INK4 locus; POR, Cytochrome P450 Oxidoreductase; MYLIP, Myosin Regulatory Light Chain Interacting Protein; HMGCR, β-hydroxy-β-methylglutaryl Coenzyme A Reductase; E, Epsilon; SLCO1B1, solute carrier organic anion transporter 1B1.
In addition, FH patients with a null mutation in the LDLR gene were identified as having a higher prevalence of CVD than those with a defective mutation [14][40][42][53]. Although these individuals at major risk of CVD are on aggressive anti-lipid regimens, most of them did not achieve the therapeutic goals of LDL-C [37][42]. On the contrary, a study by Vohl and colleagues found that the proportion of patients who achieved LDL-C targets was higher in the null mutants than in the defective mutants [37]. Schaefer et al. have confirmed that LDLR p.W556R SNP in homozygote FH patients lead to HMGCR blockers resistance but can obtain a 15% decrease of LDL-C by ezetimibe treatment. Conversely, the same LDLR mutation in patients with heterozygote FH can decrease 60% of cholesterols under a combination of ezetimibe and simvastatin [43]. These outcomes suggest that altering the LDLR should be a new pharmacological target in controlling FH.
Pharmacogenomic assays have shown that low-activity variants of HMGCR, which encode the cholesterol synthesis speed-limiting factor, can restrict the therapeutic potency of HMGCR blockers depending on the patients’ gender. For instance, the HMGCR polymorphism, rs3846662, selectively modulates women’s sensitivity to statin treatments [49]. Variations in the encoding genes of ApoA molecules and lipoprotein (A) (LPA), have been believed to constrain LDL-C response to statins and intensify coronary artery disorders [54]. Several GWAS studies have proved an association between PCSK9 polymorphisms and statin efficacy. The rs17111584 C allele in PCSK9 decreased the rosuvastatin efficacy [55], while the rs11599147 polymorphism was linked to elevated anti-lipid response [56]. A polymorphism in the WD repeat domain 52 (WDR52, rs13064411A>G) can indirectly reduce the LDLR response to statins. This mutation is associated with statin-induced elevation of PCSK9 levels that accelerate the degradation of LDLR, resulting in elevated total cholesterol levels [57]. The myosin regulatory light chain interaction protein (MYLIP) is responsible for regulating the LDLR function in cellular lipid uptake. A study noted that heterozygous FH patients with the MYLIP rs9370867 allele respond differently to statin therapy with ezetimibe based on the mutation type. After a year of treatment, the recommended cholesterol levels could be achieved in FH patients with no mutations but not in those with defective and null phenotypes [47]. All in all, the results from various studies point out to an essential role for the LDLR mutation type in predicting response to statins but also to a preponderant role to genes involved in LDLR regulation as potential modifiers to this response
References
- Khachadurian, A.K. The Inheritance of Essential Familial Hypercholesterolemia. Am. J. Med. 1964, 37, 402–407.
- Lui, D.T.W.; Lee, A.C.H.; Tan, K.C.B. Management of Familial Hypercholesterolemia: Current Status and Future Perspectives. J. Endocr. Soc. 2021, 5, bvaa122.
- Hayat, M.; Kerr, R.; Bentley, A.R.; Rotimi, C.N.; Raal, F.J.; Ramsay, M. Genetic associations between serum low LDL-cholesterol levels and variants in LDLR, APOB, PCSK9 and LDLRAP1 in African populations. PLoS ONE 2020, 15, e0229098, Correction in 2021, 16, e0249478.
- Fahed, A.C.; Nemer, G.M. Familial hypercholesterolemia: The lipids or the genes? Nutr. Metab. 2011, 8, 23.
- Galema-Boers, A.M.; Lenzen, M.J.; Engelkes, S.R.; Sijbrands, E.J.; Roeters van Lennep, J.E. Cardiovascular risk in patients with familial hypercholesterolemia using optimal lipid-lowering therapy. J Clin. Lipidol. 2018, 12, 409–416.
- Corrigendum to: 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 4255.
- Kamar, A.; Khalil, A.; Nemer, G. The Digenic Causality in Familial Hypercholesterolemia: Revising the Genotype-Phenotype Correlations of the Disease. Front. Genet. 2020, 11, 572045.
- Borges, J.B.; Oliveira, V.F.; Ferreira, G.M.; Los, B.; Barbosa, T.; Marcal, E.; Dagli-Hernandez, C.; de Freitas, R.C.C.; Bortolin, R.H.; Mori, A.A.; et al. Genomics, epigenomics and pharmacogenomics of familial hypercholesterolemia (FHBGEP): A study protocol. Res. Soc. Adm. Pharm. 2021, 17, 1347–1355.
- Chahine, J.; Kreykes, S.; Van’t Hof, J.R.; Duprez, D.; Nijjar, P. Variable and Severe Phenotypic Expression of the “Lebanese Allele” in Two Sisters with Familial Hypercholesterolemia. Vasc. Health Risk Manag. 2021, 17, 415–419.
- Fahed, A.C.; Bitar, F.F.; Khalaf, R.I.; Moubarak, E.M.; Azar, S.T.; Nemer, G.M. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine 2012, 42, 445–448.
- Hartgers, M.L.; Besseling, J.; Stroes, E.S.; Wittekoek, J.; Rutten, J.H.W.; de Graaf, J.; Visseren, F.L.J.; Imholz, B.P.M.; Roeters van Lennep, J.E.; Huijgen, R.; et al. Achieved LDL cholesterol levels in patients with heterozygous familial hypercholesterolemia: A model that explores the efficacy of conventional and novel lipid-lowering therapy. J. Clin. Lipidol. 2018, 12, 972–980.e1.
- Marziliano, N.; Medoro, A.; Mignogna, D.; Saccon, G.; Folzani, S.; Reverberi, C.; Russo, C.; Intrieri, M. Sudden Cardiac Death Caused by a Fatal Association of Hypertrophic Cardiomyopathy (MYH7, p.Arg719Trp), Heterozygous Familial Hypercholesterolemia (LDLR, p.Gly343Lys) and SARS-CoV-2 B.1.1.7 Infection. Diagnostics 2021, 11, 1229.
- Luirink, I.K.; Wiegman, A.; Kusters, D.M.; Hof, M.H.; Groothoff, J.W.; de Groot, E.; Kastelein, J.J.P.; Hutten, B.A. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N. Engl. J. Med. 2019, 381, 1547–1556.
- Perez de Isla, L.; Alonso, R.; Watts, G.F.; Mata, N.; Saltijeral Cerezo, A.; Muniz, O.; Fuentes, F.; Diaz-Diaz, J.L.; de Andres, R.; Zambon, D.; et al. Attainment of LDL-Cholesterol Treatment Goals in Patients With Familial Hypercholesterolemia: 5-Year SAFEHEART Registry Follow-Up. J. Am. Coll. Cardiol. 2016, 67, 1278–1285.
- Thedrez, A.; Blom, D.J.; Ramin-Mangata, S.; Blanchard, V.; Croyal, M.; Chemello, K.; Nativel, B.; Pichelin, M.; Cariou, B.; Bourane, S.; et al. Homozygous Familial Hypercholesterolemia Patients With Identical Mutations Variably Express the LDLR (Low-Density Lipoprotein Receptor): Implications for the Efficacy of Evolocumab. Arter. Thromb. Vasc. Biol. 2018, 38, 592–598.
- Fahed, A.C.; El-Hage-Sleiman, A.K.; Farhat, T.I.; Nemer, G.M. Diet, genetics, and disease: A focus on the middle East and north Africa region. J. Nutr. Metab. 2012, 2012, 109037.
- Ramaswami, U.; Futema, M.; Bogsrud, M.P.; Holven, K.B.; Roeters van Lennep, J.; Wiegman, A.; Descamps, O.S.; Vrablik, M.; Freiberger, T.; Dieplinger, H.; et al. Comparison of the characteristics at diagnosis and treatment of children with heterozygous familial hypercholesterolaemia (FH) from eight European countries. Atherosclerosis 2020, 292, 178–187.
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.S.; Psaty, B.M.; et al. Genomewide Association Study of Statin-Induced Myopathy in Patients Recruited Using the UK Clinical Practice Research Datalink. Clin. Pharm. Ther. 2019, 106, 1353–1361.
- Jeenah, M.; September, W.; Graadt van Roggen, F.; de Villiers, W.; Seftel, H.; Marais, D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia. Atherosclerosis 1993, 98, 51–58.
- Kajinami, K.; Yagi, K.; Higashikata, T.; Inazu, A.; Koizumi, J.; Mabuchi, H. Low-density lipoprotein receptor genotype-dependent response to cholesterol lowering by combined pravastatin and cholestyramine in familial hypercholesterolemia. Am. J. Cardiol. 1998, 82, 113–117.
- Couture, P.; Brun, L.D.; Szots, F.; Lelievre, M.; Gaudet, D.; Despres, J.P.; Simard, J.; Lupien, P.J.; Gagne, C. Association of specific LDL receptor gene mutations with differential plasma lipoprotein response to simvastatin in young French Canadians with heterozygous familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 1998, 18, 1007–1012.
- Salazar, L.A.; Cavalli, S.A.; Hirata, M.H.; Diament, J.; Forti, N.; Giannini, S.D.; Nakandakare, E.R.; Bertolami, M.C.; Hirata, R.D. Polymorphisms of the low-density lipoprotein receptor gene in Brazilian individuals with heterozygous familial hypercholesterolemia. Braz. J. Med. Biol. Res. 2000, 33, 1301–1304.
- Leitersdorf, E.; Eisenberg, S.; Eliav, O.; Friedlander, Y.; Berkman, N.; Dann, E.J.; Landsberger, D.; Sehayek, E.; Meiner, V.; Wurm, M.; et al. Genetic determinants of responsiveness to the HMG-CoA reductase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation 1993, 87, III35-44.
- Miltiadous, G.; Xenophontos, S.; Bairaktari, E.; Ganotakis, M.; Cariolou, M.; Elisaf, M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharm. Genom. 2005, 15, 219–225.
- Polisecki, E.; Muallem, H.; Maeda, N.; Peter, I.; Robertson, M.; McMahon, A.D.; Ford, I.; Packard, C.; Shepherd, J.; Jukema, J.W.; et al. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis 2008, 200, 109–114.
- Muallem, H.; North, K.E.; Kakoki, M.; Wojczynski, M.K.; Li, X.; Grove, M.; Boerwinkle, E.; Wilhelmsen, K.C.; Heiss, G.; Maeda, N. Quantitative effects of common genetic variations in the 3′UTR of the human LDL-receptor gene and their associations with plasma lipid levels in the Atherosclerosis Risk in Communities study. Hum. Genet. 2007, 121, 421–431.
- Mangravite, L.M.; Medina, M.W.; Cui, J.; Pressman, S.; Smith, J.D.; Rieder, M.J.; Guo, X.; Nickerson, D.A.; Rotter, J.I.; Krauss, R.M. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arter. Thromb. Vasc. Biol. 2010, 30, 1485–1492.
- Lahoz, C.; Pena, R.; Mostaza, J.M.; Laguna, F.; Garcia-Iglesias, M.F.; Taboada, M.; Pinto, X. Baseline levels of low-density lipoprotein cholesterol and lipoprotein (a) and the AvaII polymorphism of the low-density lipoprotein receptor gene influence the response of low-density lipoprotein cholesterol to pravastatin treatment. Metabolism 2005, 54, 741–747.
- Awan, Z.A.; Rashidi, O.M.; Al-Shehri, B.A.; Jamil, K.; Elango, R.; Al-Aama, J.Y.; Hegele, R.A.; Banaganapalli, B.; Shaik, N.A. Saudi Familial Hypercholesterolemia Patients With Rare LDLR Stop Gain Variant Showed Variable Clinical Phenotype and Resistance to Multiple Drug Regimen. Front. Med. 2021, 8, 694668.
- Santos, P.C.; Morgan, A.C.; Jannes, C.E.; Turolla, L.; Krieger, J.E.; Santos, R.D.; Pereira, A.C. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2014, 233, 206–210.
- Heath, K.E.; Gudnason, V.; Humphries, S.E.; Seed, M. The type of mutation in the low density lipoprotein receptor gene influences the cholesterol-lowering response of the HMG-CoA reductase inhibitor simvastatin in patients with heterozygous familial hypercholesterolaemia. Atherosclerosis 1999, 143, 41–54.
- Chaves, F.J.; Real, J.T.; Garcia-Garcia, A.B.; Civera, M.; Armengod, M.E.; Ascaso, J.F.; Carmena, R. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: Influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J. Clin. Endocrinol. Metab. 2001, 86, 4926–4932.
- Sun, X.M.; Patel, D.D.; Knight, B.L.; Soutar, A.K. Influence of genotype at the low density lipoprotein (LDL) receptor gene locus on the clinical phenotype and response to lipid-lowering drug therapy in heterozygous familial hypercholesterolaemia. The Familial Hypercholesterolaemia Regression Study Group. Atherosclerosis 1998, 136, 175–185.
- Sanna, C.; Stephenne, X.; Revencu, N.; Smets, F.; Sassolas, A.; Di Filippo, M.; Descamps, O.S.; Sokal, E.M. Homozygous familial hypercholesterolemia in childhood: Genotype-phenotype description, established therapies and perspectives. Atherosclerosis 2016, 247, 97–104.
- Carmena, R.; Roederer, G.; Mailloux, H.; Lussier-Cacan, S.; Davignon, J. The response to lovastatin treatment in patients with heterozygous familial hypercholesterolemia is modulated by apolipoprotein E polymorphism. Metabolism 1993, 42, 895–901.
- O’Neill, F.H.; Patel, D.D.; Knight, B.L.; Neuwirth, C.K.; Bourbon, M.; Soutar, A.K.; Taylor, G.W.; Thompson, G.R.; Naoumova, R.P. Determinants of variable response to statin treatment in patients with refractory familial hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2001, 21, 832–837.
- Vohl, M.C.; Szots, F.; Lelievre, M.; Lupien, P.J.; Bergeron, J.; Gagne, C.; Couture, P. Influence of LDL receptor gene mutation and apo E polymorphism on lipoprotein response to simvastatin treatment among adolescents with heterozygous familial hypercholesterolemia. Atherosclerosis 2002, 160, 361–368.
- Garcia-Garcia, A.B.; Gonzalez, C.; Real, J.T.; Martin de Llano, J.J.; Gonzalez-Albert, V.; Civera, M.; Chaves, F.J.; Ascaso, J.F.; Carmena, R. Influence of microsomal triglyceride transfer protein promoter polymorphism -493 GT on fasting plasma triglyceride values and interaction with treatment response to atorvastatin in subjects with heterozygous familial hypercholesterolaemia. Pharm. Genom. 2005, 15, 211–218.
- Bercovich, D.; Friedlander, Y.; Korem, S.; Houminer, A.; Hoffman, A.; Kleinberg, L.; Shochat, C.; Leitersdorf, E.; Meiner, V. The association of common SNPs and haplotypes in the CETP and MDR1 genes with lipids response to fluvastatin in familial hypercholesterolemia. Atherosclerosis 2006, 185, 97–107.
- Alonso, R.; Mata, N.; Castillo, S.; Fuentes, F.; Saenz, P.; Muniz, O.; Galiana, J.; Figueras, R.; Diaz, J.L.; Gomez-Enterria, P.; et al. Cardiovascular disease in familial hypercholesterolaemia: Influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008, 200, 315–321.
- Hu, M.; Lui, S.S.; Mak, V.W.; Chu, T.T.; Lee, V.W.; Poon, E.W.; Tsui, T.K.; Ko, G.T.; Baum, L.; Tam, L.S.; et al. Pharmacogenetic analysis of lipid responses to rosuvastatin in Chinese patients. Pharm. Genom. 2010, 20, 634–637.
- Mata, N.; Alonso, R.; Badimon, L.; Padro, T.; Fuentes, F.; Muniz, O.; Perez-Jimenez, F.; Lopez-Miranda, J.; Diaz, J.L.; Vidal, J.I.; et al. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis 2011, 10, 94.
- Schaefer, J.R.; Kurt, B.; Sattler, A.; Klaus, G.; Soufi, M. Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R). Clin. Res. Cardiol. Suppl. 2012, 7, 2–6.
- Rosales, A.; Alvear, M.; Cuevas, A.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Identification of pharmacogenetic predictors of lipid-lowering response to atorvastatin in Chilean subjects with hypercholesterolemia. Clin. Chim. Acta 2012, 413, 495–501.
- Ahmed, W.; Ali, I.S.; Riaz, M.; Younas, A.; Sadeque, A.; Niazi, A.K.; Niazi, S.H.; Ali, S.H.; Azam, M.; Qamar, R. Association of ANRIL polymorphism (rs1333049:C>G) with myocardial infarction and its pharmacogenomic role in hypercholesterolemia. Gene 2013, 515, 416–420.
- Drogari, E.; Ragia, G.; Mollaki, V.; Elens, L.; Van Schaik, R.H.; Manolopoulos, V.G. POR*28 SNP is associated with lipid response to atorvastatin in children and adolescents with familial hypercholesterolemia. Pharmacogenomics 2014, 15, 1963–1972.
- Santos, P.C.; Morgan, A.C.; Jannes, C.E.; Krieger, J.E.; Santos, R.D.; Pereira, A.C. The MYLIP p.N342S polymorphism is associated with response to lipid-lowering therapy in Brazilian patients with familial hypercholesterolemia. Pharm. Genom. 2014, 24, 548–555.
- Mabuchi, H.; Nohara, A.; Noguchi, T.; Kobayashi, J.; Kawashiri, M.A.; Inoue, T.; Mori, M.; Tada, H.; Nakanishi, C.; Yagi, K.; et al. Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis 2014, 236, 54–61.
- Leduc, V.; Bourque, L.; Poirier, J.; Dufour, R. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharm. Genom. 2016, 26, 1–11.
- Fazio, S.; Robertson, D.G.; Joh, T.; Wan, H.; Riel, T.; Forgues, P.; Baum, C.M.; Garzone, P.D.; Gumbiner, B. Effects of 12 weeks of treatment with intravenously administered bococizumab, a humanized monoclonal antibody blocking proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects on high-dose statin. Cardiovasc Ther. 2018, 36, e12308.
- Bea, A.M.; Lamiquiz-Moneo, I.; Marco-Benedi, V.; Mateo-Gallego, R.; Perez-Calahorra, S.; Jarauta, E.; Martin, C.; Cenarro, A.; Civeira, F. Lipid-lowering response in subjects with the p.(Leu167del) mutation in the APOE gene. Atherosclerosis 2019, 282, 143–147.
- Dagli-Hernandez, C.; de Freitas, R.C.C.; Marcal, E.; Goncalves, R.M.; Faludi, A.A.; Borges, J.B.; Bastos, G.M.; Los, B.; Mori, A.A.; Bortolin, R.H.; et al. Late response to rosuvastatin and statin-related myalgia due to SLCO1B1, SLCO1B3, ABCB11, and CYP3A5 variants in a patient with Familial Hypercholesterolemia: A case report. Ann. Transl. Med. 2021, 9, 76.
- Charland, S.L.; Agatep, B.C.; Herrera, V.; Schrader, B.; Frueh, F.W.; Ryvkin, M.; Shabbeer, J.; Devlin, J.J.; Superko, H.R.; Stanek, E.J. Providing patients with pharmacogenetic test results affects adherence to statin therapy: Results of the Additional KIF6 Risk Offers Better Adherence to Statins (AKROBATS) trial. Pharm. J. 2014, 14, 272–280.
- Paquette, M.; Bernard, S.; Thanassoulis, G.; Baass, A. LPA genotype is associated with premature cardiovascular disease in familial hypercholesterolemia. J. Clin. Lipidol. 2019, 13, 627–633.e1.
- Chasman, D.I.; Giulianini, F.; MacFadyen, J.; Barratt, B.J.; Nyberg, F.; Ridker, P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012, 5, 257–264.
- Thompson, J.F.; Hyde, C.L.; Wood, L.S.; Paciga, S.A.; Hinds, D.A.; Cox, D.R.; Hovingh, G.K.; Kastelein, J.J. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ. Cardiovasc. Genet. 2009, 2, 173–181.
- de Keyser, C.E.; Becker, M.L.; Hofman, A.; Lous, J.J.; Uitterlinden, A.G.; Visser, L.E.; Stricker, B.H. The rs13064411 polymorphism in the WDR52 gene, associated with PCSK9 levels, modifies statin-induced changes in serum total and LDL cholesterol levels. Pharm. Genom. 2015, 25, 134–142.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
711
Revisions:
5 times
(View History)
Update Date:
11 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No