Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yi Zhang | + 1603 word(s) | 1603 | 2021-09-09 04:47:45 | | | |
| 2 | Camila Xu | + 310 word(s) | 1913 | 2021-09-30 11:07:22 | | | | |
| 3 | Camila Xu | + 310 word(s) | 1913 | 2021-09-30 11:08:24 | | | | |
| 4 | Conner Chen | Meta information modification | 1913 | 2021-10-12 05:15:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, Y. Aberrant Stress Granule Dynamics. Encyclopedia. Available online: https://encyclopedia.pub/entry/14772 (accessed on 07 February 2026).
Zhang Y. Aberrant Stress Granule Dynamics. Encyclopedia. Available at: https://encyclopedia.pub/entry/14772. Accessed February 07, 2026.
Zhang, Yi. "Aberrant Stress Granule Dynamics" Encyclopedia, https://encyclopedia.pub/entry/14772 (accessed February 07, 2026).
Zhang, Y. (2021, September 30). Aberrant Stress Granule Dynamics. In Encyclopedia. https://encyclopedia.pub/entry/14772
Zhang, Yi. "Aberrant Stress Granule Dynamics." Encyclopedia. Web. 30 September, 2021.
Copy Citation
Stress granules are membrane-less organelles formed through the process of liquid–liquid phase separation (LLPS) under certain stress conditions, such as oxidative stress and heat shock, among others.
stress granule
aggrephagy
neurodegenerative disease
amyotrophic lateral sclerosis
phase separation
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive degeneration of the upper and lower motor neurons, resulting in a loss of motor function and eventually death. About 10% of ALS cases are familial (fALS), while about 90% are sporadic (sALS). Identification of ALS-causative genes, including superoxide dismutase 1 (SOD1), transactive response DNA-binding protein 43 (TARDBP-43), fused in sarcoma (FUS), chromosome 9 open reading frame 72 (C9orf72), heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), valosin-containing protein (VCP), ubiquilin 2 (UBQLN2), sequestosome 1 (SQSTM1/p62), annexin A11 (ANXA11), optineurin (OPTN), and TANK (TRAF-associated NF-κB activator)-binding kinase 1 (TBK1) have advanced the understanding of ALS pathogenesis. ALS gene products are considered resident stress granule (SG) components or SG-associated proteins (Table 1) [1].
Table 1. The representative ALS-causative proteins.
| Protein | Associated NDDs | Important Structures | Function | Main Pathogenesis | Role in SG Dynamics | References |
|---|---|---|---|---|---|---|
| TDP-43 | FTD, ALS, PD, HD | C-terminal Glycine-rich domain, RNA recognition motifs (RRM1 and RRM2), nuclear localization signal and nuclear export signal | Regulates mRNA splicing, translation, transportation and stability | Mutations cause loss of TDP-43 nuclear function and cytoplasmic accumulation | SG component | Reviewed in [2] |
| FUS | FTD, ALS | N terminal prion-like domain, RNA recognition motif, C terminal nuclear localization signal | RNA- binding protein aids RNA transcription and splicing | Mutations on NLS impair the FUS nuclear transport causing cytoplasmic aggregation | SG component | Reviewed in [3] |
| C9ORF72 | ALS, FTD | - | Affect transcription, translation and RNA transport |
Abnormal hexanucleotide GGGGCC repeat amplification | Cause stress and interact with SG proteins | Reviewed in [4] |
| SOD1 | ALS | - | An antioxidant enzyme detoxifying superoxide | Mutated SOD1 exposes hydrophobic surfaces and N-terminal short region increasing aggregation propensity | Cause stress and interact with SG proteins | Reviewed in [5] |
| UBQLN2 | ALS, FTD | Ubiquitin-like domain (UBL), UBA, four stress-induced protein 1-like domains (STI-1 like), PXX domain | Directs misfolded or redundant proteins to the proteasome, acts in macroautophagy | Missense mutations | SG autophagic clearance | Reviewed in [6] |
| ANXA11 | ALS | Four conserved annexin (ANX) domains, low-complexity domain (LCD) | Regulates cytokinesis, vesicle trafficking, apoptosis, intracellular Ca2+ homeostasis and stress granule dynamics | Missense mutations | Cause stress and interact with SG proteins | Reviewed in [7] |
| VCP | FTD, ALS | N-terminal domain, ATP-binding domains D1 and D2 | DNA damage response, cell cycle control, autophagy, and SG clearance | Mutations disrupt the autophagic degradation of ubiquitinated proteins, resulting in the accumulation of non-degradative autophagosomes | SG component; SG autophagic clearance | Reviewed in [8] |
| MATR3 | ALS, FTD, AD | Two tandem RNA-recognition motifs, two zinc finger domains | Alternative splicing, mRNA stability, transcription and mRNA nuclear export | Missense mutations | SG component | Reviewed in [9] |
NDD: neurodegenerative disease; PD: Parkinson’s disease; AD: Alzheimer’s disease; HD: Huntington’s disease; ALS: amyotrophic lateral sclerosis; FTD: frontotemporal dementia.
Stress granules are membrane-less organelles formed through the process of liquid–liquid phase separation (LLPS) under certain stress conditions, such as oxidative stress and heat shock, among others [1][10]. SGs are transient cellular compartments that undergo dynamic assembly and dissociation. However, chronic stress can lead to persistent stress granules, eventually resulting in the aggregation of disease-related proteins.
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved lysosomal degradative pathway, which is essential in the cellular and organismal levels of homeostasis [11][12][13]. Morphologically, autophagy is initiated by the formation of phagophores in mammalian cells. After nucleation of the phagophore, the membrane expands to generate an autophagosome, which fuses with a lysosome or vacuole, leading to the degradation of the cargo [14][15][16][17]. Clearance of the cytosolic components, such as protein aggregates, is conferred by cargo receptors that specifically recognize the cargo [18][19][20][21][22][23][24][25]. Dysfunction of autophagy is highly associated with various human diseases [26][27][28], such as neurodegenerative diseases.
Protein aggregates derived from interrupted SG dynamics pose a toxic insult, which can be partially mitigated by a selective autophagy pathway called aggrephagy (Figure 1). Aggresomes formed by those insoluble protein aggregates and labeled by ubiquitins are considered to initiate the process of aggrephagy. Aggresomes are transported to a microtubule-organizing center with the help of histone deacetylase 6 (HDAC6), which binds to ubiquitinated cargos [29]. Aggrephagy is controlled by a panel of receptor proteins, such as p62, next to BRCA1 gene 1 (NBR1), toll interacting protein (TOLLIP), OPTN, and Tax1 binding protein 1 (TAXBP1) [30]. Mechanistically, these receptors bridge ubiquitinated protein aggregates with autophagosomal membranes by simultaneously binding to ubiquitin chains and the lipidated LC3-family proteins via ubiquitin-associated (UBA) domains and LC3 interacting region (LIR) motifs, respectively [22][23]. These receptors are able to work both independently and cooperatively. For instance, NBR1 can interact with p62 and promote its phase separation [31]. Autophagy-linked FYVE-domain containing protein (ALFY) interacts with p62 and binds to several autophagy-related proteins, playing a role in the formation of autophagic membranes. The fusion between aggresomes and lysosomes involves proteins including Rab7, marking the final degradation of protein aggregates [29]. It should be noted that the mutations of two of these aggrephagy receptors, p62 and OPTN, are implicated in ALS.
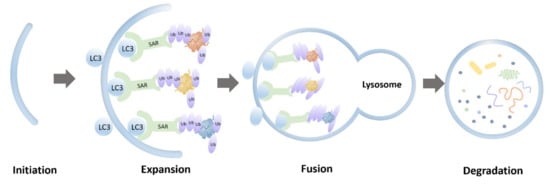
Figure 1. Schematic representation of aggrephagy.
Loss of SG homeostasis and defective aggrephagy are common pathological features of neurogenerative diseases [1][31][32][33][34][35][36]. VCP is encoded by an ALS causal gene and is a critical regulator mediating autophagic degradation of abnormal stress granules [37]. In this review, we will discuss the intersection of aggrephagy and stress granules in the pathogenesis of ALS.
2. Superoxide Dismutase 1 (SOD1)
Superoxide dismutase 1 gene encoding Cu/Zn superoxide dismutase was the first identified ALS-related gene [38]. The enzyme protects cells by detoxifying superoxide radicals O2−. SOD1 gene mutations account for approximately 20% of fALS. Although no consensus linking SOD1 mutations to toxicity has been reached [35], it is generally accepted that ubiquitinated cytoplasmic inclusions formed by ALS-causing SOD1 mutants contribute to toxicity in ALS [35]. Most ALS-associated mutations significantly impact the immature states of SOD1, destabilizing the metal-free and disulfide-reduced polypeptide, which leads to unfolding at physiological temperatures [39]. Moreover, mutations that change the hydrophobicity of SOD1 or cause cellular Ca2+ dysregulation promote the aggregation tendency of SOD1 mutants in ALS [40][41]. In addition, T cell-restricted intracellular antigen 1 (TIA-1) positive SGs can alter the dynamics of stress granules [42].
SOD1 is not a resident protein of SGs. However, mutant SOD1 interacts with TIA-1, one of the core components of stress granules associated with ALS. Mutant SOD1 increases the number of TIA-1 positive SGs. The abnormal interaction between mutant SOD1 and TIA-1 alters the dynamic of stress granules [42]. In addition, mutant SOD1 binds to GTPase-activating protein-(SH3 domain)-binding protein 1 (G3BP1), another protein marker of SGs, in an RNA-independent manner interfering with the dynamics of G3BP1-positive SGs [43]. Therefore, these findings suggest that aberrant interactions between SOD1, TIA-1, and G3BP1 might dysregulate SG.
Mutant SOD1 aggregates can be recognized by p62 and targeted for autophagic degradation [44][45]. Furthermore, mutant SOD1 aggregates may sequester OPTN, resulting in a reduced mitophagy flux, accounting for neurodegeneration [46]. However, whether the perturbation of SGs dynamics by SOD1 mutants impacts protein aggregation tendency remains unclear. Further studies are needed to confirm the exact role of aggrephagy in SOD1-associated ALS and the specific aggrephagy receptors involved [47].
3. Transactive Response DNA-Binding Protein 43 (TDP-43)
Transactive response DNA-binding protein 43 belongs to the heterogeneous ribonucleoprotein family. TDP-43 plays a critical role in diverse cellular processes, such as regulating RNA splicing, pre-microRNA processing, messenger RNA transport, and stress granule formation [48]. Hyper-phosphorylated, ubiquitinated, and cleaved TDP-43 aggregation has been identified as a pathological protein in disease-affected central nervous system regions [49]. Furthermore, TDP-43 has been detected as abnormal cytoplasmic aggregates in neurons and glia of more than 90% of ALS and 45% of frontotemporal dementia (FTD) cases [50].
TDP-43 can aggregate and propagate in a seed-dependent, self-templating, prion-like manner in vitro and in vivo [51]. Under chronic cell stress, TDP-43 is recruited to the cytoplasmic SGs, which evolve to form insoluble pathological aggregates [52][53]. TDP-43 also interacts with the four other ALS causal gene products, HNRNPA1, HNRNPA2B1, matrin 3 (MATR3), and UBQLN2 [54][55][56][57], which are resident proteins in SGs. The identification of TIA-1 as an ALS causal gene further reinforces the fact that TDP-43 in ALS is formed via altered LLPS [58]. These observations suggest that many ALS causal genes may converge on the TDP-43 pathway associated with pathologies.
Several studies have confirmed that autophagy plays a role in clearing TDP-43 aggregates. Significant colocalization between selective autophagy receptor p62 with TPD-43 aggregates was observed in ALS/FTD, indicating that the autophagy pathway could prevent the accumulation of TDP-43 aggregates [59]. In addition, VCP and OPTN appear to colocalize with TDP-43 inclusions in the spinal motor neurons of ALS patients [60]. Upregulation of autophagy leads to reduced TDP-43 proteinopathy in the nervous system of ALS/FTD transgenic mice models, which further validates the role of autophagy in mitigating toxicity of TDP-43 mutants [61][62]. Conversely, TDP-43 also plays a role in the regulation of autophagy by binding to ATG7 mRNA via RNA recognition motif 1(RRM1). Down-regulation of TDP-43 decreases ATG7 mRNA levels, which abolishes autophagosome expansion [63]. Furthermore, the loss of TDP-43 impairs the fusion of autophagosomes with lysosomes through decreasing dynactin 1, a component of the dynein-dynactin complex involved in lysosome transportation. The impaired fusion finally leads to the accumulation of immature autophagic vesicles blocking the autophagy-lysosome pathway [64].
4. Fused in Sarcoma (FUS)
FUS was first discovered in 1993 as a fusion oncogene in human liposarcoma located on chromosome 16 [65][66]. It contains 15 exons encoding a 526-amino acid protein. Moreover, it contains an N-terminal Gln-Gly-Ser-Tyr (QGSY)-rich domain, an RNA-recognition motif, three Arg-Gly-Gly repeat domains (RGG1-3), a zinc-finger motif and a C-terminal nuclear localization signal (NLS) [67]. In 2009, pathological inclusion bodies containing mutant FUS protein were recognized in fALS6 cases [68][69]. Approximately 2/3 of FUS mutations are located on exons 12–15, which encode zinc-finger motif, RGG2 and RGG3 domains, and the NLS. Other mutations are located on exons 3–6, encoding QGSY-rich and RGG1 domains. The C-terminal mutations are twice as likely to occur in fALS than in sALS, while mutations within exons 3–6 are more common in sALS. C-terminal ALS mutations are pathological, as they disrupt NLS [70][71]. They cause defective nuclear import of FUS and cytoplasmic mislocalization. Cytoplasmic FUS mislocalization leads to nuclear loss of function and triggers motor neuron death through a toxic gain of function [72].
Arginine residues in RGG motifs are required for phase separation of FUS. Loss of FUS arginine methylation promotes phase separation and SG association of FUS [73]. Prion-like domains of FUS are located on the QGSY-rich and C-terminal RGG2 domain, contributing to FUS phase separation and aggregation. ALS-associated FUS mutants can bind and sequester wild type (WT) FUS into cytoplasmic SGs [74], accelerating aberrant liquid to solid phase transition of stress granules [75]. The nuclear import receptor (NIR), also known as Transportin-1, recognizes the NLS domain; therefore, it chaperons FUS from the cytoplasm to the nucleus. NIRs can reverse aberrant phase separation and aggregation of proteins with prion-like domains, including FUS and TDP-43, to mitigate neurodegeneration in vivo [73][76].
R521C and P525L are two common FUS mutations associated with ALS. FUS-R521C causes DNA damage and RNA splicing defects [77]. It colocalizes with stress granules, significantly increasing SG assembly and persistence [78]. FUS-R521C-positive SGs were colocalized to LC3-positive autophagosomes accumulating in autophagy-deficient neurons, suggesting that autophagy is involved in the clearance of FUS mutants [79]. P525L FUS mutation causes early-onset of ALS [80]. P525L-positive SGs are more intense and larger than the WT. The PI3K/AKT/mTOR pathway inhibition increases autophagy by reducing FUS recruitment into SGs and reduces abnormal SGs linked to P525L FUS [81]. Accumulation of ubiquitinated proteins and autophagy receptor p62 was detected in neuronal cells with ALS-associated FUS mutation due to impaired autophagy [82]. However, overexpression of Rab1 rescued these defects, suggesting that Rab1 has a protective role in ALS [83].
References
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666.
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20.
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348.
- Xu, X.F.; Su, Y.; Zou, Z.Y.; Zhou, Y.L.; Yan, J.G. Correlation between C9ORF72 mutation and neurodegenerative diseases: A comprehensive review of the literature. Int. J. Med. Sci. 2021, 18, 378–386.
- Hayashi, Y.; Homma, K.; Ichijo, H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul. 2016, 60, 95–104.
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103.
- Nahm, M.; Lim, S.M.; Kim, Y.E.; Park, J.; Noh, M.Y.; Lee, S.; Roh, J.E.; Hwang, S.M.; Park, C.K.; Kim, Y.H.; et al. ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci. Transl. Med. 2020, 12, eaax3993.
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. p97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J. Med. Chem. 2020, 63, 1892–1907.
- Malik, A.M.; Barmada, S.J. Matrin 3 in neuromuscular disease: Physiology and pathophysiology. JCI Insight 2021, 6, e143948.
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 2020, 370, 56–60.
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93.
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467.
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635.
- Bernard, A.; Klionsky, D.J. Autophagosome formation: Tracing the source. Dev. Cell 2013, 25, 116–117.
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233.
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757.
- Moreau, K.; Ravikumar, B.; Renna, M.; Puri, C.; Rubinsztein, D.C. Autophagosome precursor maturation requires homotypic fusion. Cell 2011, 146, 303–317.
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156.
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178.
- Farre, J.C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552.
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724.
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16.
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242.
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22.
- Kirkin, V. History of the Selective Autophagy Research: How Did It Begin and Where Does It Stand Today? J. Mol. Biol. 2019, 432, 3–27.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42.
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28.
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285.
- Sun, D.; Wu, R.; Li, P.; Yu, L. Phase Separation in Regulation of Aggrephagy. J. Mol. Biol. 2020, 432, 160–169.
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200.
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40.
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842.
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62.
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274.
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: A link to toxic effects of calcium overload in amyotrophic lateral sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228.
- Tompa, D.R.; Kadhirvel, S. Changes in hydrophobicity mainly promotes the aggregation tendency of ALS associated SOD1 mutants. Int. J. Biol. Macromol. 2020, 145, 904–913.
- Lee, D.Y.; Jeon, G.S.; Sung, J.J. ALS-Linked Mutant SOD1 Associates with TIA-1 and Alters Stress Granule Dynamics. Neurochem. Res. 2020, 45, 2884–2893.
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576.
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073.
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417.
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525.
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303.
- Afroz, T.; Perez-Berlanga, M.; Polymenidou, M. Structural Transition, Function and Dysfunction of TDP-43 in Neurodegenerative Diseases. CHIMIA Int. J. Chem. 2019, 73, 380–390.
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- Nonaka, T.; Hasegawa, M. TDP-43 Prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024463.
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e337.
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051.
- Gilpin, K.M.; Chang, L.; Monteiro, M.J. ALS-linked mutations in ubiquilin-2 or hnRNPA1 reduce interaction between ubiquilin-2 and hnRNPA1. Hum. Mol. Genet. 2015, 24, 2565–2577.
- Cassel, J.A.; Reitz, A.B. Ubiquilin-2 (UBQLN2) binds with high affinity to the C-terminal region of TDP-43 and modulates TDP-43 levels in H4 cells: Characterization of inhibition by nucleic acids and 4-aminoquinolines. Biochim. Biophys. Acta 2013, 1834, 964–971.
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473.
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666.
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e809.
- Hiji, M.; Takahashi, T.; Fukuba, H.; Yamashita, H.; Kohriyama, T.; Matsumoto, M. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008, 116, 183–191.
- Ayaki, T.; Ito, H.; Fukushima, H.; Inoue, T.; Kondo, T.; Ikemoto, A.; Asano, T.; Shodai, A.; Fujita, T.; Fukui, S.; et al. Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathol. Commun. 2014, 2, 172.
- Kumar, S.; Phaneuf, D.; Cordeau, P., Jr.; Boutej, H.; Kriz, J.; Julien, J.P. Induction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD. Mol. Neurodegener. 2021, 16, 1.
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685.
- Bose, J.K.; Huang, C.C.; Shen, C.K. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448.
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142.
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644.
- Rabbitts, T.H.; Forster, A.; Larson, R.; Nathan, P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993, 4, 175–180.
- Iko, Y.; Kodama, T.S.; Kasai, N.; Oyama, T.; Morita, E.H.; Muto, T.; Okumura, M.; Fujii, R.; Takumi, T.; Tate, S.; et al. Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 2004, 279, 44834–44840.
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208.
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211.
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275.
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857.
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterle, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097.
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e713.
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobagyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol. Genet. 2013, 22, 2676–2688.
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077.
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692.e620.
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999.
- Acosta, J.R.; Goldsbury, C.; Winnick, C.; Badrock, A.P.; Fraser, S.T.; Laird, A.S.; Hall, T.E.; Don, E.K.; Fifita, J.A.; Blair, I.P.; et al. Mutant Human FUS Is Ubiquitously Mislocalized and Generates Persistent Stress Granules in Primary Cultured Transgenic Zebrafish Cells. PLoS ONE 2014, 9, e90572.
- Ryu, H.H.; Jun, M.H.; Min, K.J.; Jang, D.J.; Lee, Y.S.; Kim, H.K.; Lee, J.A. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol. Aging 2014, 35, 2822–2831.
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75.
- Marrone, L.; Poser, I.; Casci, I.; Japtok, J.; Reinhardt, P.; Janosch, A.; Andree, C.; Lee, H.O.; Moebius, C.; Koerner, E.; et al. Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell Rep. 2018, 10, 375–389.
- Ho, W.Y.; Ling, S.C. Elevated FUS levels by overriding its autoregulation produce gain-of-toxicity properties that disrupt protein and RNA homeostasis. Autophagy 2019, 15, 1665–1667.
- Soo, K.Y.; Sultana, J.; King, A.E.; Atkinson, R.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.; Farg, M.A.; Atkin, J.D. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015, 1, 15030.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
975
Revisions:
4 times
(View History)
Update Date:
12 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No