 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrick Ming Kuen Tang | + 776 word(s) | 776 | 2020-06-08 05:25:37 | | | |
| 2 | Nicole Yin | + 74 word(s) | 850 | 2020-06-09 10:44:32 | | | | |
| 3 | Nicole Yin | -3 word(s) | 847 | 2020-11-06 04:14:37 | | |
Video Upload Options
Emerging studies suggest that unsolved inflammation will progressively transit into kidney fibrosis that finally results in an irreversible end-stage renal disease (ESRD). Increasing studies have suggested pathogenic roles of innate immunity in the kidney diseases. A better understanding of the underlying mechanisms may uncover a novel therapeutic strategy for ESRD.
1. Introduction
Renal injury eventually progresses to chronic kidney diseases (CKD) under unresolved inflammation[1]. Upon kidney injury, damage-associated molecular patterns (DAMPs) trigger inflammatory responses, resulting in immunocytes infiltration predominantly to neutrophils, macrophages, and natural killer cells[2](Figure 1). Together with resident dendritic cells, innate immune cells thus take corresponding roles in damage and repair on the site of injury. Recent studies reveal that renal cell death releases endogenous cytokines, chemokines, oxidative stress, and DAMPs, which largely promotes infiltration and activation of immune cells that result in CKD[3][4][5][6].
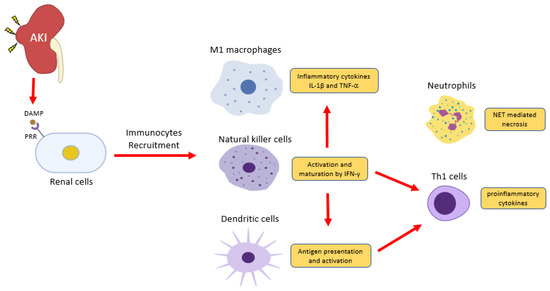
Figure 1. Inflammatory cells in acute kidney injury (AKI). Renal cells damage releases damage-associated molecular patterns (DAMPs) to induce inflammation via pattern recognition receptors (PRRs). Natural killer cells release IFN-γ to induce classical activation of macrophage (M1)- producing pro-inflammatory cytokines IL-1 and TNF2 and induce dendritic cell maturation for pro-inflammatory Th1 differentiation. DAMPs also trigger the release of a neutrophils extracellular trap (NET) to further recruit inflammatory cells and induce renal cell death.
2. Neutrophils
Inflammation triggers the production of reactive oxygen species (ROS) and serine proteases of neutrophils upon adhesion to the injured site, which are believed to combat bacterial infection[3] and trigger the formation of neutrophil extracellular traps (NETs)[4]. NETs are found in acute tubular necrosis as a unique form of cell death, where intracellular membranes are degraded due to the histones and granule proteins attached on the ejecting chromatin[5]. Chromatin-released NETs also act as DAMPs to elicit inflammatory and cytotoxic effects, and Singh et al. reported that the histones released from NETs could enter and kill renal cells by nonspecific DNA and RNA binding. Besides, extracellular histones can ligate to the toll-like receptors ‒2 and ‒4 and nucleotide-binding domain (NOD)-like receptor protein 3 for inducing inflammasome[6]. Altogether, NETs generate an auto-amplification loop of inflammation to accelerate tubular necrosis, therefore causing irreversible damage to the nephrons[7].
3. Dendritic Cells
DCs are antigen-presenting cells. They are derived from bone marrow as an immature state as precursor DCs, then circulate into peripheral blood for foreign and pathogenic antigens detection[8]. Upon inflammatory stimuli at the injured site, endogenous DAMPs and PAMPs trigger the maturation of DCs for antigen presentation, cytokines, and co-stimulatory molecules expression. Interestingly, inflammatory stimuli transform dendritic cells to be a distinct subset called inflammatory DCs (infDCs), which activate T cells for promoting inflammation[9][10]. The infDCs secrete IL-1, TNF-ɑ, IL-12, and IL-23 to stimulate IL-17 production in CD4+ and CD8+ T cells in vivo[10]. Inhibition of Flt3, a ligand for cross-presentation between DCs and T cells, significantly reduces infiltration and proliferation of CD4+ and CD8+ T cells, therefore alleviating kidney inflammation in experimental adriamycin nephropathy models[11]. These findings suggest DCs could activate adaptive immunity and the autoimmune response for facilitating renal inflammation.
4. Natural Killer Cells
Natural killer (NK) cells are modulators of innate and adaptive immune responses. Their surface activating and inhibitory receptors are responsible for regulating NK cells’ activities upon interactions to target cells, complementary and antagonist pathways that are initiated to trigger NK cells to secrete cytokines and chemokines to regulate neighboring immune cells[12][13]. Studies found that NK cells could promote Th1 polarization of CD4+ T cells and maturation of DCs through IFN-γ[14]. In addition, the activated NK cells are capable of eliminating DCs that fail to complete their maturation[15]. It is believed that NK cells modulate differential immune responses depending on the cytokine environment. Recent in vitro studies found that exposure of NK cells to exogenous IL-12 would induce strong cytolytic activity against immature DCs; in contrast, IL-4-conditioned NK cells would generate DCs favoring T cell polarization or Th2 priming[16]. Thus, NK cells regulate DCs and T cells in the renal microenvironment.
5. Macrophages
Macrophages are highly plastic, and they contribute to every stage of CKD, from renal inflammation to fibrosis. In experimental CKD models, endogenous DAMPs and PAMPs induce M1 pro-inflammatory macrophages[17][18][19][20], therefore producing inflammatory cytokines IL-1β and TNFa to promote renal inflammation[21][22][23]. Nevertheless, studies also reported that macrophage infiltration correlates with active fibrotic lesions, supported by the significant reduction in renal fibrosis in IRI and UUO models under macrophage depletion[24][25]. M1 macrophages induce chronic renal inflammation, resulting in collagen and extracellular matrix deposition[26]. During CKD progression, M1 is gradually replaced by the reparative M2 phenotype[27]. The M1/M2 transition is evident and characterized by a time-dependent exchange of M1/M2 markers and the existence of their intermediate population, detected by single-cell sequencing analysis in AKI, glomerular disease, and UUO models[28][29][30]. Clinical studies of diabetic nephropathy[31] and kidney transplantation[32] showed that M2 macrophages localize at the fibrotic areas and actively produce pro-fibrotic molecules IL-1, PDGF, MMP-2/9/12, and galectin 3[33][34]. Moreover, bone marrow-derived macrophages (BMDM) could further differentiate into ɑ-SMA+ myofibroblasts locally in injured kidney under unresolved inflammation via macrophage-myofibroblast transition (MMT) for promoting renal fibrosis[35][32]. These studies demonstrate the pro-fibrotic role of M2 macrophages in renal fibrosis.
References
- Yohei Ikezumi; Lyn Hurst; Robert C. Atkins; David J. Nikolic-Paterson; Macrophage-mediated renal injury is dependent on signaling via the JNK pathway.. Journal of the American Society of Nephrology 2004, 15, 1775-1784, 10.1097/01.asn.0000131272.06958.de.
- Kate Schroder; Matthew J. Sweet; D A Hume; Signal integration between IFNγ and TLR signalling pathways in macrophages. Immunobiology 2006, 211, 511-524, 10.1016/j.imbio.2006.05.007.
- Christina C. N. Wu; JongDae Lee; Eyal Raz; Maripat Corr; Dennis A. Carson; Necessity of Oligonucleotide Aggregation for Toll-like Receptor 9 Activation. Journal of Biological Chemistry 2004, 279, 33071-33078, 10.1074/jbc.m311662200.
- Mi Ryu; Onkar P. Kulkarni; Ewa Radomska; Nicolai Miosge; Oliver Gross; Hans-Joachim Anders; Bacterial CpG-DNA accelerates Alport glomerulosclerosis by inducing an M1 macrophage phenotype and tumor necrosis factor-α-mediated podocyte loss. Kidney International 2011, 79, 189-198, 10.1038/ki.2010.373.
- N I Tomosugi; S J Cashman; H Hay; C D Pusey; D J Evans; A Shaw; A J Rees; Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor, and IL-1.. The Journal of Immunology 1989, 142, 3083–3090.
- Jennifer R. Timoshanko; Jonathon D. Sedgwick; Stephen R. Holdsworth; Peter G. Tipping; Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis.. Journal of the American Society of Nephrology 2003, 14, 1785-1793, 10.1097/01.asn.0000073902.38428.33.
- Vinicius Andrade-Oliveira; Orestes Foresto-Neto; Ingrid Kazue Mizuno Watanabe; Roberto Zatz; Niels Olsen Saraiva Camara; Inflammation in Renal Diseases: New and Old Players.. Frontiers in Pharmacology 2019, 10, 1192, 10.3389/fphar.2019.01192.
- Gregory H Tesch; N Yang; H Yu; H Y Lan; R Foti; Sj Chadban; R C Atkins; David J Nikolic-Paterson; Intrinsic renal cells are the major source of interleukin-1 beta synthesis in normal and diseased rat kidney.. Nephrology Dialysis Transplantation 1997, 12, 1109–1115.
- Yingjie Han; Frank Y Ma; Greg H Tesch; Carl L Manthey; David J. Nikolic-Paterson; c-fms blockade reverses glomerular macrophage infiltration and halts development of crescentic anti-GBM glomerulonephritis in the rat. Laboratory Investigation 2011, 91, 978-991, 10.1038/labinvest.2011.61.
- Myung-Gyu Kim; Sun Chul Kim; Yoon Sook Ko; Hee Young Lee; Sang Kyung Jo; Wonyong Cho; The Role of M2 Macrophages in the Progression of Chronic Kidney Disease following Acute Kidney Injury. PLOS ONE 2015, 10, e0143961, 10.1371/journal.pone.0143961.
- Zhen Liu; Xiao Ru Huang; Hai-Yong Chen; Josef M. Penninger; Hui-Yao Lan; Loss of angiotensin-converting enzyme 2 enhances TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation in a mouse model of obstructive nephropathy. Laboratory Investigation 2012, 92, 650-661, 10.1038/labinvest.2012.2.
- Bing Shen; Xiuheng Liu; Yu Fan; Jianxin Qiu; Macrophages Regulate Renal Fibrosis Through Modulating TGFβ Superfamily Signaling. Inflammation 2014, 37, 2076-2084, 10.1007/s10753-014-9941-y.
- Don C Rockey; P. Darwin Bell; Joseph A. Hill; Fibrosis — A Common Pathway to Organ Injury and Failure. New England Journal of Medicine 2015, 372, 1138-1149, 10.1056/nejmra1300575.
- Sarah Huen; Lloyd G. Cantley; Macrophage-mediated injury and repair after ischemic kidney injury.. Pediatric Nephrology 2014, 30, 199-209, 10.1007/s00467-013-2726-y.
- Ling Lin; Kebin Hu; Tissue-type plasminogen activator modulates macrophage M2 to M1 phenotypic change through annexin A2-mediated NF-κB pathway. Oncotarget 2017, 8, 88094-88103, 10.18632/oncotarget.21510.
- Yingjie Han; Frank Y. Ma; Greg H. Tesch; Carl L. Manthey; David J. Nikolic-Paterson; Role of macrophages in the fibrotic phase of rat crescentic glomerulonephritis. American Journal of Physiology-Renal Physiology 2013, 304, F1043-F1053, 10.1152/ajprenal.00389.2012.
- Julie Belliere; Audrey Casemayou; Laure Ducasse; Alexia Zakaroff-Girard; Frédéric Martins; Jason S. Iacovoni; Céline Frugier; Bénédicte Buffin-Meyer; Bernard Pipy; Minique Chauveau; et al.Joost P. SchanstraJean Loup Bascands Specific Macrophage Subtypes Influence the Progression of Rhabdomyolysis-Induced Kidney Injury. Journal of the American Society of Nephrology 2014, 26, 1363-1377, 10.1681/ASN.2014040320.
- Celine Q.F. Klessens; Malu Zandbergen; Ron Wolterbeek; Jan A. Bruijn; Ton J. Rabelink; Ingeborg M. Bajema; Daphne Ijpelaar; Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrology Dialysis Transplantation 2016, 32, 1322–1329, 10.1093/ndt/gfw260.
- Yohei Ikezumi; Toshiaki Suzuki; Takeshi Yamada; Hiroya Hasegawa; Utako Kaneko; Masanori Hara; Toshio Yanagihara; David J. Nikolic-Paterson; Akihiko Saitoh; Alternatively activated macrophages in the pathogenesis of chronic kidney allograft injury. Pediatric Nephrology 2014, 30, 1007-1017, 10.1007/s00467-014-3023-0.
- Xuanyi Du; Akira Shimizu; Yukinari Masuda; Naomi Kuwahara; Takashi Arai; Mitue Kataoka; Masaaki Uchiyama; Tomohiro Kaneko; Toshio Akimoto; Yasuhiko Iino; et al.Yuh Fukuda Involvement of matrix metalloproteinase-2 in the development of renal interstitial fibrosis in mouse obstructive nephropathy. Laboratory Investigation 2012, 92, 1149-1160, 10.1038/labinvest.2012.68.
- Brian Czaya; Christian Faul; FGF23 and inflammation-a vicious coalition in CKD.. Kidney International 2019, 96, 813-815, 10.1016/j.kint.2019.05.018.
- Giuseppe Cianciolo; Andrea Galassi; Irene Capelli; Roberto Schillaci; Gaetano La Manna; Mario Cozzolino; Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White?. Current Vascular Pharmacology 2018, 16, 143-156, 10.2174/1570161115666170310092202.
- Neil C. Henderson; Alison C. MacKinnon; Sarah L. Farnworth; Tiina Kipari; Christopher Haslett; John P. Iredale; Fu-Tong Liu; Jeremy Hughes; Tariq Sethi; Galectin-3 Expression and Secretion Links Macrophages to the Promotion of Renal Fibrosis. The American Journal of Pathology 2008, 172, 288-298, 10.2353/ajpath.2008.070726.
- Shuang Wang; Xiao-Ming Meng; Yee-Yung Ng; Frank Y. Ma; Shuang Zhou; Yang Zhang; Chen Yang; Xiao-Ru Huang; Jun Xiao; Ying-Ying Wang; et al.Shuk-Man KaYong-Jiang TangArthur C.K. ChungKa-Fai ToDavid J. Nikolic-PatersonHui-Yao Lan TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2015, 7, 8809-8822, 10.18632/oncotarget.6604.
- Franco Klingberg; Boris Hinz; Eric S. White; The myofibroblast matrix: implications for tissue repair and fibrosis.. The Journal of Pathology 2013, 229, 298-309, 10.1002/path.4104.
- Ying-Ying Wang; Hong Jiang; Jun Pan; Xiao-Ru Huang; Yu-Cheng Wang; Hong-Feng Huang; Ka-Fai To; David J. Nikolic-Paterson; Hui-Yao Lan; Jiang-Hua Chen; et al. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. Journal of the American Society of Nephrology 2017, 28, 2053-2067, 10.1681/ASN.2016050573.
- Valerie S. LeBleu; Gangadhar Taduri; Joyce O'connell; Yingqi Teng; Vesselina G. Cooke; Craig Woda; Hikaru Sugimoto; Raghu Kalluri; Origin and function of myofibroblasts in kidney fibrosis. Nature Medicine 2013, 19, 1047-1053, 10.1038/nm.3218.
- Masaki Fujimoto; Yoshiro Maezawa; Koutaro Yokote; Kensuke Joh; Kazuki Kobayashi; Harukiyo Kawamura; Motonobu Nishimura; Anita B. Roberts; Yasushi Saito; Seijiro Mori; et al. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy.. Biochemical and Biophysical Research Communications 2003, 305, 1002-1007, 10.1016/s0006-291x(03)00885-4.
- J.-A. Moon; H.-T. Kim; I.-S. Cho; Y.Y. Sheen; Dae-Kee Kim; IN-1130, a novel transforming growth factor-β type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney International 2006, 70, 1234-1243, 10.1038/sj.ki.5001775.
- Jun Wang; Shougang Zhuang; Src family kinases in chronic kidney disease.. American Journal of Physiology-Renal Physiology 2017, 313, F721-F728, 10.1152/ajprenal.00141.2017.
- Patrick Tang; Shuang Zhou; Chun-Jie Li; Jinyue Liao; Jun Xiao; Qing-Ming Wang; Guang-Yu Lian; Jinhong Li; Xiao-Ru Huang; Ka-Fai To; et al.Alexander Kc LeungCharing C. N. ChongRonald C. MaTin-Lap LeeHui-Yao Lan The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney International 2018, 93, 173-187, 10.1016/j.kint.2017.07.026.
- A. C. Wiseman; Immunosuppressive Medications.. Clinical Journal of the American Society of Nephrology 2015, 11, 332-343, 10.2215/CJN.08570814.
- Deborah Mattinzoli; Masami Ikehata; Koji Tsugawa; Carlo Maria Alfieri; Paola Dongiovanni; Elena Trombetta; Luca Valenti; A. Puliti; Lorenza Lazzari; Piergiorgio Messa; et al. FGF23 and Fetuin-A Interaction in the Liver and in the Circulation. International Journal of Biological Sciences 2018, 14, 586-598, 10.7150/ijbs.23256.
- Rupal Mehta; Xuan Cai; Jungwha Lee; Dawei Xie; Xue Wang; Julia Scialla; Amanda H. Anderson; Jon Taliercio; Mirela Dobre; Jing Chen; et al.Michael FischerMary LeonardJames LashChi-Yuan HsuIan H. De BoerHarold I. FeldmanMyles WolfTamara IsakovaLawrence J. AppelAlan S. GoJiang HePanduranga S. RaoMahboob RahmanRaymond R. TownsendCRIC Study Investigators Serial Fibroblast Growth Factor 23 Measurements and Risk of Requirement for Kidney Replacement Therapy: The CRIC (Chronic Renal Insufficiency Cohort) Study. American Journal of Kidney Diseases 2020, 75, 908-918, 10.1053/j.ajkd.2019.09.009.
- Taro Kawai; Shizuo Akira; The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology 2010, 11, 373-384, 10.1038/ni.1863.


