Emerging studies suggest that unsolved inflammation will progressively transit into kidney fibrosis that finally results in an irreversible end-stage renal disease (ESRD). Increasing studies have suggested pathogenic roles of innate immunity in the kidney diseases. A better understanding of the underlying mechanisms may uncover a novel therapeutic strategy for ESRD.
- chronic kidney disease
- immunity
- inflammation
1. Introduction
Renal injury eventually progresses to chronic kidney diseases (CKD) under unresolved inflammation[1] [104]. Upon kidney injury, damage-associated molecular patterns (DAMPs) trigger inflammatory responses, resulting in immunocytes infiltration predominantly to neutrophils, macrophages, and natural killer cells[2] [105] (Figure 1). Together with resident dendritic cells, innate immune cells thus take corresponding roles in damage and repair on the site of injury. Recent studies reveal that renal cell death releases endogenous cytokines, chemokines, oxidative stress, and DAMPs, which largely promotes infiltration and activation of immune cells that result in CKD[3][4][5][6].
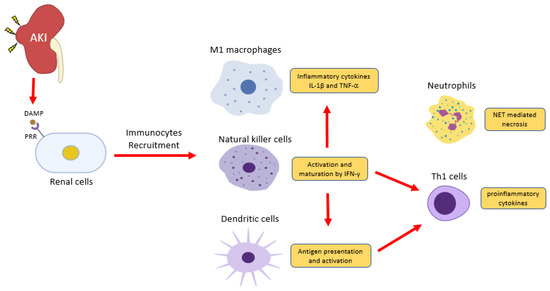
Figure 1. Inflammatory cells in acute kidney injury (AKI). Renal cells damage releases damage-associated molecular patterns (DAMPs) to induce inflammation via pattern recognition receptors (PRRs). Natural killer cells release IFN-γ to induce classical activation of macrophage (M[1)- producing pro-inflammatory cytokines IL-1 and TNF2 and induce dendritic cell maturation for pro-inflammatory Th1 differentiation06‒109]. DAMPs also trigger the release of a neutrophils extracellular trap (NET) to further recruit inflammatory cells and induce renal cell death.
2. Neutrophils
Inflammation triggers the production of reactive oxygen species (ROS) and serine proteases of neutrophils upon adhesion to the injured site, which are believed to combat bacterial infection[3] [106] and trigger the formation of neutrophil extracellular traps (NETs)[4] [107]. NETs are found in acute tubular necrosis as a unique form of cell death, where intracellular membranes are degraded due to the histones and granule proteins attached on the ejecting chromatin[5] [108]. Chromatin-released NETs also act as DAMPs to elicit inflammatory and cytotoxic effects, and Singh et al. reported that the histones released from NETs could enter and kill renal cells by nonspecific DNA and RNA binding. Besides, extracellular histones can ligate to the toll-like receptors ‒2 and ‒4 and nucleotide-binding domain (NOD)-like receptor protein 3 for inducing inflammasome[6] [109]. Altogether, NETs generate an auto-amplification loop of inflammation to accelerate tubular necrosis, therefore causing irreversible damage to the nephrons[7] [18].
3. Dendritic Cells
DCs are antigen-presenting cells. They are derived from bone marrow as an immature state as precursor DCs, then circulate into peripheral blood for foreign and pathogenic antigens detection[8] [110]. Upon inflammatory stimuli at the injured site, endogenous DAMPs and PAMPs trigger the maturation of DCs for antigen presentation, cytokines, and co-stimulatory molecules expression. Interestingly, inflammatory stimuli transform dendritic cells to be a distinct subset called inflammatory DCs (infDCs), which activate T cells for promoting inflammation[9][10] [111, 112]. The infDCs secrete IL-1, TNF-ɑ, IL-12, and IL-23 to stimulate IL-17 production in CD4+ and CD8+ T cells in vivo[10] [112]. Inhibition of Flt3, a ligand for cross-presentation between DCs and T cells, significantly reduces infiltration and proliferation of CD4+ and CD8+ T cells, therefore alleviating kidney inflammation in experimental adriamycin nephropathy models[11] [22]. These findings suggest DCs could activate adaptive immunity and the autoimmune response for facilitating renal inflammation.
4. Natural Killer Cells
Natural killer (NK) cells are modulators of innate and adaptive immune responses. Their surface activating and inhibitory receptors are responsible for regulating NK cells’ activities upon interactions to target cells, complementary and antagonist pathways that are initiated to trigger NK cells to secrete cytokines and chemokines to regulate neighboring immune cells[12][13] [113, 114]. Studies found that NK cells could promote Th1 polarization of CD4+ T cells and maturation of DCs through IFN-γ[14] [115]. In addition, the activated NK cells are capable of eliminating DCs that fail to complete their maturation[15] [116]. It is believed that NK cells modulate differential immune responses depending on the cytokine environment. Recent in vitro studies found that exposure of NK cells to exogenous IL-12 would induce strong cytolytic activity against immature DCs; in contrast, IL-4-conditioned NK cells would generate DCs favoring T cell polarization or Th2 priming[16] [117]. Thus, NK cells regulate DCs and T cells in the renal microenvironment.
5. Macrophages
Macrophages are highly plastic, and they contribute to every stage of CKD, from renal inflammation to fibrosis. In experimental CKD models, endogenous DAMPs and PAMPs induce M1 pro-inflammatory macrophages[17][18][19][20] [118‒121], therefore producing inflammatory cytokines IL-1β and TNFa to promote renal inflammation[21][22][23] [25,26,122]. Nevertheless, studies also reported that macrophage infiltration correlates with active fibrotic lesions, supported by the significant reduction in renal fibrosis in IRI and UUO models under macrophage depletion[24][25] [123, 124]. M1 macrophages induce chronic renal inflammation, resulting in collagen and extracellular matrix deposition[26] [125]. During CKD progression, M1 is gradually replaced by the reparative M2 phenotype[27] [126]. The M1/M2 transition is evident and characterized by a time-dependent exchange of M1/M2 markers and the existence of their intermediate population, detected by single-cell sequencing analysis in AKI, glomerular disease, and UUO models[28][29][30] [127‒129]. Clinical studies of diabetic nephropathy[31] [130] and kidney transplantation[32] [131] showed that M2 macrophages localize at the fibrotic areas and actively produce pro-fibrotic molecules IL-1, PDGF, MMP-2/9/12, and galectin 3[33][34] [27, 28]. Moreover, bone marrow-derived macrophages (BMDM) could further differentiate into ɑ-SMA+ myofibroblasts locally in injured kidney under unresolved inflammation via macrophage-myofibroblast transition (MMT) for promoting renal fibrosis[35][32] [31, 131]. These studies demonstrate the pro-fibrotic role of M2 macrophages in renal fibrosis.