Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yang Liu | + 2289 word(s) | 2289 | 2021-09-18 18:05:13 | | | |
| 2 | Conner Chen | Meta information modification | 2289 | 2021-10-08 07:58:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Liu, Y. D-2-Hydroxyglutarate in Glioma Biology. Encyclopedia. Available online: https://encyclopedia.pub/entry/14629 (accessed on 07 February 2026).
Liu Y. D-2-Hydroxyglutarate in Glioma Biology. Encyclopedia. Available at: https://encyclopedia.pub/entry/14629. Accessed February 07, 2026.
Liu, Yang. "D-2-Hydroxyglutarate in Glioma Biology" Encyclopedia, https://encyclopedia.pub/entry/14629 (accessed February 07, 2026).
Liu, Y. (2021, September 27). D-2-Hydroxyglutarate in Glioma Biology. In Encyclopedia. https://encyclopedia.pub/entry/14629
Liu, Yang. "D-2-Hydroxyglutarate in Glioma Biology." Encyclopedia. Web. 27 September, 2021.
Copy Citation
Oncometabolites, the abnormally accumulated metabolites derived from disrupted cancer metabolic pathways, are a recently defined concept in cancer biology. Isocitrate dehydrogenase (IDH) mutations are common genetic abnormalities in glioma, which result in the accumulation of an “oncometabolite”, D-2-hydroxyglutarate (D-2-HG). Abnormally elevated D-2-HG levels result in a distinctive pattern in cancer biology, through competitively inhibiting α-ketoglutarate (α-KG)/Fe(II)-dependent dioxgenases (α-KGDDs). D-2-HG affects DNA/histone methylation, hypoxia signaling, DNA repair, and redox homeostasis, which impacts the oncogenesis of IDH-mutated cancers.
glioma
oncometabolites
IDH1/2mut
D-2-HG
epigenetic
DDR
1. Metabolism and Oncometabolites
Metabolites refer to the intermediate or end products of the metabolic pathways that are involved in cell growth, development, and survival [1][2]. The distinctive pattern of cancer metabolism was first described by the German physiologist Otto H. Warburg in the 1920s, who proposed that tumor cells exhibit remarkably high glucose consumption compared to non-malignant tissues [3][4]. Cancer cells prefer glucose consumption via aerobic glycolysis, which is 10–100 times faster than mitochondria respiration, and renders an overall benefit to cell proliferation [5]. This preference for aerobic glycolysis was later named the Warburg effect, which highlights the distinctive metabolic pathways in cancer cells [6].
The discovery of oncometabolites extends the understanding of the unique metabolic routes in cancer cells. Oncometabolites are abnormally accumulated metabolites that are involved in various critical aspects throughout cancer progression [7]. In contrast to adaptive metabolic reprogramming, the production of oncometabolites commonly results from genetic abnormalities in the genes encoding critical metabolic products. Succinate, fumarate, D-2-HG, and L-2-HG are considered oncometabolites [8].
2. Cancer-Associated IDH Mutation and D-2-HG
2-hydroxyglutarate (2-HG) is a metabolite detected in urine that was first described by Karl Heinrich Ritthausen in 1868 [9]. In 1980, Chalmers and Duran identified two similar neurometabolic disorder types related to 2-HG, L-2-hydroxyglutaric aciduria (L-2-HGA) [10] and D-2-hydroxyglutaric aciduria (D-2-HGA) [11]. Mutations in L-2-hydroxyglutarate dehydrogenase and D-2-hydroxyglutarate dehydrogenase (D2HGDH) result in the manifestations of L-2-HGA and D-2-HGA, respectively [12]. Mutations in the mitochondrial citrate carrier SLC25A1 cause combined D-2- and L-2-HGA. Interestingly, the study pointed out half of the patients with D-2-HGA lack the D2HGDH mutation but instead carried mutations in IDH2 [13]. On the other hand, IDH mutations result in the biosynthesis of D-2-HG from α-ketoglutarate. As mentioned above, somatic mutations in IDH have been identified in glioma and other human malignancies through genome-wide mutation analysis [14][15]. To date, cancer-associated IDH1/2 mutations are commonly found in acute myeloid leukemia (~20%) [16], melanoma [17], cartilaginous tumors (56–70%) [18], cholangiocarcinoma (8.5–20%) [19], and WHO II/III gliomas (~80%) [20][21]. There are three IDH isoforms in mammalian cells: one cytosolic form (IDH1) and two mitochondrial forms (IDH2 and IDH3). IDH1 and IDH2 are homodimers, which consume nicotinamide adenine dinucleotide phosphate (NADP+) for their catalytic function. IDH3 is a heterotetramer and is a nicotinamide adenine dinucleotide (NAD+)-dependent enzyme. IDH1/2 functions as β-decarboxylating dehydrogenases, which can reversibly convert isocitrate to α-ketoglutarate (α-KG), an essential metabolic intermediate in the Krebs cycle that regulates metabolic and catalytic processes [22]. Heterozygous IDH1/2 mutations frequently occur in the arginine residues of the catalytic pockets IDH1 (R132H) and IDH2 (R140Q, R172K) [23][24]. These IDH1/2 mutations alter the organization of the catalytic centers in these enzymes, which establish gain-of-function changes in their catalytic function, as well as the production of D-2-HG (Figure 1) [23][25][26][27]. The chemical structure of D-2-HG is similar to α-KG. The only difference is the carbonyl group in the C2 position that is replaced by the hydroxyl group [28]. Therefore, D-2-HG could interfere with the enzymes that employ α-KG as a substrate, and competitively inhibit α-KGDDs by occupying the α-KG binding sites in the enzyme [29]. Moreover, α-KGDDs are a highly diversified enzyme family that is involved in many critical biological processes, such as DNA/histone demethylation, ubiquitination, and hydroxylation, and regulate epigenetic alternation, protein stability, and different signaling (e.g., HIF-1 and mTOR) [30][31].
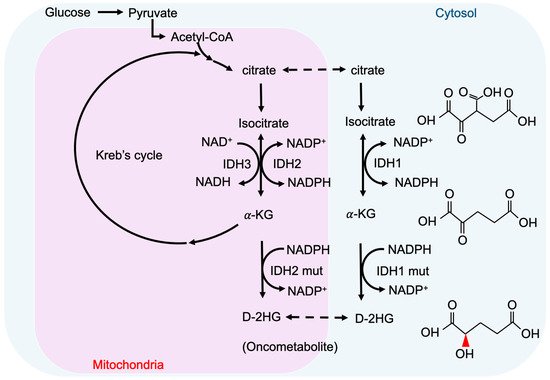
Figure 1. IDH mutations and production of D-2-HG. IDH1 and IDH2 are NADP+ dependent enzymes and distribute in the cytosol and mitochondria, respectively. IDH3 is a NAD+ dependent enzyme that locates in mitochondria. Mutations of IDH1 and IDH2 enzymes are sufficient to convert a-KG to D-2HG. NADP, nicotinamide adenine dinucleotide phosphate, NADPH, the reduced form of NADP.
Identifying IDH mutants and their subtypes is a common strategy for molecular pathology in glioma diagnosis. It mainly relies on immunohistochemistry, DNA sequencing, and measurements of intratumoral and circulating D-2-HG, which rely on mass spectrometry (MS)-based [32][33], enzymatic assay-based, and magnetic resonance (MR)-based methods [34]. To precisely detect the levels of D-2-HG in gliomas with an MS-based platform, several studies suggested collecting samples from patients’ cerebrospinal fluid instead of from serum, as CSF has higher D-2-HG concentrations and provides more specific results [35]. Furthermore, the MS-based method could not clearly distinguish L- and D-2HG, which requires the use of additional chiral derivatization to separate these enantiomers. Although the MS and assay-based methods provide relatively high sensitivity (~ μM), circulating samples must be collected invasively and the D-2-HG final concentration cannot directly reflect the actual tumor size and border. Several recent studies discovered that non-invasive diagnostic approaches, such as magnetic resonance (MR)-based imaging (MRI) [36] and spectroscopy (MRS) [37] could be used to predict IDH mutations by measuring D-2-HG in gliomas with mM level sensitivity. Magnetic resonance spectroscopic imaging (MRSI) integrates the information of MRS and MRI, which can detect and quantify various metabolites, and generate the metabolite map from multiple lesions within the brain [38], which adds value to conventional MRI in pre-operation planning and post-treatment monitoring. MRS-based methods provide 88.6% accuracy in identifying the IDH mutational status, with 89.5% sensitivity and 81.3% specificity, suggesting that MR-based techniques are safe and promising approaches to support glioma diagnosis [39].
3. Epigenetic Regulation by D-2-HG
High concentrations of D-2-HG are needed to competitively bind to various α-KGDDs [40], which include a vast spectrum of demethylases, such as ten-eleven translocation enzymes (TETs) [29], the Jumonji (JmjC) domain-containing lysine-specific histone demethylases (JmjC-KDMs) [41], and fat mass and obesity-associated protein (FTO) [42] (Figure 2). D-2-HG-induced DNA and histone hypermethylation have led to the aberrant expression of oncogenes and tumor suppressor genes and play a key role in malignant transformation of IDH-mutated cancers [43][44]. In addition, a high concentration of D-2-HG inhibits the demethylase function of FTO, which decreases the stability of transcripts, and results in the suppression of relevant pathways [42].
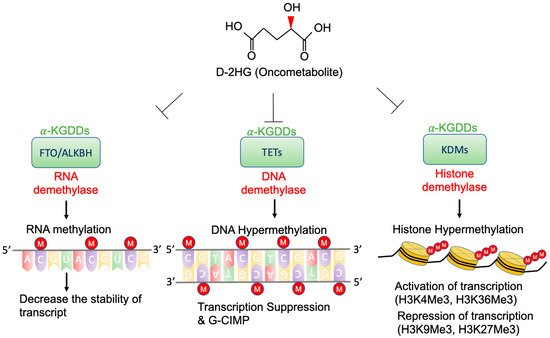
Figure 2. Epigenetic alterations of D-2-HG. D-2-HG alters the methylation status of DNA, RNA, and histone to regulate gene expression, and RNA stability via inhibition of various types of α-KDGG.
3.1. TETs and G-CIMP
DNA methylation is considered as a gene repressive mark. The levels and patterns of DNA methylation are regulated by DNA methyltransferases (DNMT) and TETs [45]. The TET family contains three members (TET1, 2, and 3) [46], and the primary function is to catalyze the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), further to 5-fluorocytosine (5-fC), and 5-carboxylcytosine (5-caC) [47]. The 5-caC is eventually decarboxylated by thymine-DNA glycosylase (TDG) and converted to cytosine. TET-mediated demethylation plays a critical role in regulation of gene expression [48], DNA base excision repair [49], and chromosome replication [50]. Experimental evidence shows that expression of IDH1mut R132H or IDH2mut R172K inhibits TET1/2 activity and decreases the level of 5-hmC [38]. Deficiency of TET2 catalytic function could lead to oncogenesis, through global hypermethylation and further enhanced cellular proliferation [51]. Although loss-of-function mutations of TET1/2 are less frequently found in glioma [52], the presence of D-2-HG in IDH mutated cancer is sufficient to block the activity of TETs, which results in genome-wide DNA hypermethylation [29][53][54]. Two major types of hypermethylation have been described: gene-specific hypermethylation in the cytosine-phosphate-guanine (CpG) island of the promoter area, and widespread (non-promoter) hypermethylation [55][56]. Hypermethylation in tumor suppressor genes has been reported to correlate to cell malignant transformation and tumorigenesis [57]. Several lines of evidence indicate that IDH-mutated gliomas exhibit a distinctive CpG islands methylation phenotype (CIMP) [58][59] through remodeling the methylome and is sufficient to change the epigenome, and further alter the transcriptional programs and the differentiation state [60]. Therefore, glioma CIMP (G-CIMP) could be used as a classification standard and diagnosis indicator. Based on the clinical observations, G-CIMP positive patients are relatively younger [58] and have more favorable outcomes than G-CIMP-patients [61]. However, not all the IDH-mutant/G-CIMP glioma patients exhibit a consistent prognosis [43]. Noushmehr et al. further categorized G-CIMP into two subgroups based on the methylation level: IDH mutant / G-CIMP-high and IDH mutant / G-CIMP-low [58], which could be considered as a novel epigenetic signature, independent of genomic and histopathologic classification criteria, to refine the diagnosis [61]. In high-grade glioma, IDH mutant / G-CIMP-high patients show more extended overall survival and favorable prognosis than IDH mutant / G-CIMP-low [60].
3.2. KDMs and Histone Methylation
Histone methylation plays a critical role in chromatin dynamics and transcriptional regulation [62]. In eukaryotes, most histone methylation occurs in the lysine and arginine residues of histone 3 and 4 (H3, H4), and serves as an epigenetic mechanism to regulate gene transcription. N(6)-methyllysine residue demethylation is regulated by two types of KDM subfamilies: flavin-dependent KDMs and JmjC-KDMs [62], and JmjC-KDMs are one of the α-KGDD members. The presence of high-level D-2-HG is sufficient to suppress the catalytic function of JmjC-KDMs, and subsequently induce global histone methylation [63][64]. In IDH1/2 mutated gliomas, the high concentration of D-2-HG could suppress the function of KDM4A, KDM4B, and KDM4C (also known as JmjC-KDM2A, JmjC-KDM2B, and JmjC-KDM2C), and increase histone methylation levels, such as H3K9me3, H3K9me2, H3K36me3, and H3K4me3 [64][65][66]. Among all these D-2-HG mediated histone/chromatin regulators, trimethylation of H3K4, H3K36, and H3K79 acts as a transcriptional activator [67][68], and trimethylation of H3K9 and H3K27 acts as a transcriptional repressor [69]. Histone methylation influences almost all biological processes and contributes to cancer initiation, progression and/or metastasis in various malignancies [70]. Several studies showed that tri-methylation of H3K4, H3K9 and H3K27 is present in IDH-mutated cancers [63][71]. However, the biological roles of the histone methylation pattern and the potential roles in glioma pathogenesis remain elusive.
3.3. FTO and RNA Methylation
FTO is a RNA N6-methyladenosine (m6A) demethylase [72], which mediates mRNA m6A modification and changes the stability of target RNAs. Su et al. discovered that high concentration D-2-HG induces cell-cycle arrest and apoptosis in D-2-HG sensitive (without IDH mutations) AML via FTO/m6A mediated MYC inhibition [42]. Interestingly in IDH IDH1/2-mutant AML, leukemia cells can tolerate this inhibitory activity. Furthermore, Qing et al. also demonstrated that D-2-HG abrogates FTO-mediated post-transcriptional upregulation of glycolytic genes and further results in suppression of aerobic glycolysis [73].
4. Signaling Pathway Alterations and D-2-HG
4.1. HIF-1 Signaling Pathway
Hypoxia-inducible factors (HIFs) are critical transcription factors that are sensitive to oxygen concentration. HIF is a heterodimer composed of the constitutively expressed HIF-1β subunit and the oxygen-regulated HIF-1α subunit [74]. Several pioneering studies have revealed the role of HIFs in critical cancer hallmarks such as oncogenesis, metabolism, and therapy resistance [67][75]. Overexpression of HIF-α has been identified in various malignancies [76], which regulates apoptosis, tumor angiogenesis, and cellular proliferation [77]. The expression level of HIF-1α is significantly associated with poor survival in patients with high-grade (III+IV) gliomas [78]. The function of HIFs is mainly regulated by their post-translational modifications. Under normoxic conditions, HIF-α is hydroxylated by prolyl hydroxylases (prolyl hydroxylases domain proteins, PHDs) and asparaginyl hydroxylase (factor inhibiting HIF, FIHs), which guide the HIF-α protein to von Hippel-Lindau (VHL) mediated proteolysis [79]. Both PHDs and FIHs are α-KGDDs, which can be affected by the presence of D-2-HG. In IDH1/2mut glioma cell lines, Zhao et al. described that a high concentration of D-2-HG suppresses the activity of PHDs and FIHs, which reduces HIF-1α degradation, and increases HIF-1-dependent transcription [80]. However, their study results were in contrast with the findings by Koivunen et al. which indicated that D-2-HG either links to activation of PHDs [81], or is insufficient to affect HIF-1 [82]. Sun et al. also demonstrated that in the IDH1 knock-in mice model, U87 glioma cell line, and clinical databases, angiogenesis-related factors, including ANGPT1, PDGFB, and VEGFA, were downregulated in the IDH-mutated gliomas group, and promoter regions were also highly hyper-methylated [83]. The contradictory evidence suggests that the molecular mechanism could be complicated regarding how D-2-HG impacts the hypoxia-sensing pathway. Further research is encouraged to further dissect the relationship between D-2-HG and the hypoxia-sensing pathways.
4.2. RTK and mTOR Signaling Pathway
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase belonging to the phosphatidylinositol 3-kinase-related kinase (PI3K) family and serves as a core protein in the mTOR complex1 (mTORC1) and the mTOR complex2 (mTORC2). mTOR is mainly activated by extracellular activators, such as insulin-like growth factor 1 (IGF1), vascular endothelial growth factor (VEGF), and epidermal growth factor receptor (EGFR). mTORC1 and mTORC2 regulate different cellular processes and play important roles in cancer cell proliferation, migration, and survival [84][85][86][87].
The mTOR pathway could be activated via D-2-HG blockade of KDM4A [88]. In addition to histone demethylation, KDM4A mediates the demethylation process of cytosolic proteins, which may affect their function and stability. The DEP domain-containing mTOR-interacting protein (DEPTOR) is an endogenous negative regulator of the mTOR pathway and widely expressed in the human brain [89]. The loss of DEPTOR could activate mTOR downstream signaling [90]. KDM4A reduces the ubiquitination of DEPTOR by non-chromatin binding, catalytic activity to suppress β-transducin repeat-containing protein 1 (β-TrCP1) ubiquitin E3 ligase, and stabilization of DEPTOR [88][91]. The presence of D-2-HG in IDH1/2 mutated gliomas induced inhibition of KDM4A, which decreases the half-life and protein level of DEPTOR, and further enhances mTORC1/2 kinase activities [88]. The activated mTORC1/2 phosphorylates S6K1, Akt, and SGK1 to promote cell growth and survival [90]. Our previous study demonstrated an alternative mechanism of mTOR activation, the expression of Rictor upregulated in IDH-mutated glioma patients’ samples and cell lines, which enhanced the mTORC1/Rac1 downstream signaling and further increased the endocytosis [92].
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359.
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38.
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519.
- Warburg, O. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137.
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218.
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314.
- Collins, R.R.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A new paradigm for oncology, metabolism, and the clinical laboratory. Clin. Chem. 2017, 63, 1812–1820.
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2, 85.
- Ritthausen, H. Ueber die glutansäure, das zersetzungsproduct der glutaminsäure durch salpetrige säure. J. Für Prakt. Chem. 1868, 103, 239–242.
- Duran, M.; Kamerling, J.; Bakker, H.; Van Gennip, A.; Wadman, S. L-2-Hydroxyglutaric aciduria: An inborn error of metabolism? J. Inherit. Metab. Dis. 1980, 3, 109–112.
- Chalmers, R.; Lawson, A.; Watts, R.; Tavill, A.; Kamerling, J.; Hey, E.; Ogilvie, D. d-2-Hydroxyglutaric aciduria: Case report and biochemical studies. J. Inherit. Metab. Dis. 1980, 3, 11–15.
- Struys, E.A.; Salomons, G.S.; Achouri, Y.; Van Schaftingen, E.; Grosso, S.; Craigen, W.J.; Verhoeven, N.M.; Jakobs, C. Mutations in the D-2-hydroxyglutarate dehydrogenase gene cause D-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2005, 76, 358–360.
- Kranendijk, M.; Struys, E.A.; Van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; Van Der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; De Klerk, J.B. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 2010, 330, 336.
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812.
- Abbas, S.; Lugthart, S.; Kavelaars, F.G.; Schelen, A.; Koenders, J.E.; Zeilemaker, A.; van Putten, W.J.; Rijneveld, A.W.; Löwenberg, B.; Valk, P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: Prevalence and prognostic value. Blood J. Am. Soc. Hematol. 2010, 116, 2122–2126.
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089.
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p. R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 2010, 163, 747–755.
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011, 43, 1262–1265.
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765.
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773.
- Juratli, T.A.; Kirsch, M.; Robel, K.; Soucek, S.; Geiger, K.; von Kummer, R.; Schackert, G.; Krex, D. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J. Neuro-Oncol. 2012, 108, 403–410.
- Cojocaru, E.; Wilding, C.; Engelman, B.; Huang, P.; Jones, R.L. Is the IDH Mutation a Good Target for Chondrosarcoma Treatment? Curr. Mol. Biol. Rep. 2020, 6, 1–9.
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234.
- Yan, H.; Bigner, D.D.; Velculescu, V.; Parsons, D.W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009, 69, 9157–9159.
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543.
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308.
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R (−)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498.
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 2015, 18, 16–26.
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30.
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015, 22, 508–515.
- Shapira, S.N.; Christofk, H.R. Metabolic Regulation of Stem Cell Fate and Function. FASEB J. 2020, 34, 1.
- Struys, E.A.; Jansen, E.E.; Verhoeven, N.M.; Jakobs, C. Measurement of urinary D-and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography–tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin. Chem. 2004, 50, 1391–1395.
- Sahm, F.; Capper, D.; Pusch, S.; Balss, J.; Koch, A.; Langhans, C.D.; Okun, J.G.; von Deimling, A. Detection of 2-hydroxyglutarate in formalin-fixed paraffin-embedded glioma specimens by gas chromatography/mass spectrometry. Brain Pathol. 2012, 22, 26–31.
- Yuan, B.-F. Quantitative Analysis of Oncometabolite 2-Hydroxyglutarate. In Cancer Metabolomics. Advances in Experimental Medicine and Biology; Hu, S., Ed.; Springer: Cham, Switzerland, 2021; Volume 1280, pp. 161–172.
- Kalinina, J.; Ahn, J.; Devi, N.S.; Wang, L.; Li, Y.; Olson, J.J.; Glantz, M.; Smith, T.; Kim, E.L.; Giese, A.; et al. Selective detection of the D-enantiomer of 2-hydroxyglutarate in the CSF of glioma patients with mutated isocitrate dehydrogenase. Clin. Cancer Res. 2016, 22, 6256–6265.
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1474.
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629.
- Laino, M.E.; Young, R.; Beal, K.; Haque, S.; Mazaheri, Y.; Corrias, G.; Bitencourt, A.G.; Karimi, S.; Thakur, S.B. Magnetic resonance spectroscopic imaging in gliomas: Clinical diagnosis and radiotherapy planning. BJR Open 2020, 2, 20190026.
- Tietze, A.; Choi, C.; Mickey, B.; Maher, E.A.; Ulhøi, B.P.; Sangill, R.; Lassen-Ramshad, Y.; Lukacova, S.; Østergaard, L.; Von Oettingen, G. Noninvasive assessment of isocitrate dehydrogenase mutation status in cerebral gliomas by magnetic resonance spectroscopy in a clinical setting. J. Neurosurg. 2017, 128, 391–398.
- Reiter-Brennan, C.; Semmler, L.; Klein, A. The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas. Contemp. Oncol. 2018, 22, 215–222.
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70.
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 2018, 172, 90–105.e23.
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563.
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72.
- Hamidi, T.; Singh, A.K.; Chen, T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics 2015, 7, 247–265.
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710.
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303.
- Bowman, R.L.; Levine, R.L. TET2 in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Med. 2017, 7, a026518.
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517.
- Mahfoudhi, E.; Talhaoui, I.; Cabagnols, X.; Della Valle, V.; Secardin, L.; Rameau, P.; Bernard, O.A.; Ishchenko, A.A.; Abbes, S.; Vainchenker, W.; et al. TET2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Repair 2016, 43, 78–88.
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843.
- Kraus, T.F.; Greiner, A.; Steinmaurer, M.; Dietinger, V.; Guibourt, V.; Kretzschmar, H.A. Genetic characterization of ten-eleven-translocation methylcytosine dioxygenase alterations in human glioma. J. Cancer 2015, 6, 832.
- Liu, Y.; Jiang, W.; Liu, J.; Zhao, S.; Xiong, J.; Mao, Y.; Wang, Y. IDH1 mutations inhibit multiple α-ketoglutarate-dependent dioxygenase activities in astroglioma. J. Neuro-Oncol. 2012, 109, 253–260.
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567.
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163.
- Duncan, C.G.; Barwick, B.G.; Jin, G.; Rago, C.; Kapoor-Vazirani, P.; Powell, D.R.; Chi, J.-T.; Bigner, D.D.; Vertino, P.M.; Yan, H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012, 22, 2339–2355.
- Wu, B.-K.; Brenner, C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014, 9, 1827–1840.
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522.
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153.
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483.
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro-Oncology 2018, 20, 608–620.
- Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816.
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469.
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478.
- Lu, C.; Venneti, S.; Akalin, A.; Fang, F.; Ward, P.S.; DeMatteo, R.G.; Intlekofer, A.M.; Chen, C.; Ye, J.; Hameed, M.; et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013, 27, 1986–1998.
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306.
- Höpfl, G.; Ogunshola, O.; Gassmann, M. HIFs and tumors—Causes and consequences. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 286, R608–R623.
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837.
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469.
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251.
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N 6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887.
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9.
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480.
- Patiar, S.; Harris, A.L. Role of hypoxia-inducible factor-1α as a cancer therapy target. Endocr.-Relat. Cancer 2006, 13, S61–S75.
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8, S62–S67.
- Carmeliet, P.; Dor, Y.; Herbert, J.-M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490.
- Liu, Q.; Cao, P. Clinical and prognostic significance of HIF-1α in glioma patients: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 22073–22083.
- Shaw, K. Environmental cues like hypoxia can trigger gene expression and cancer development. Nat. Educ. 2008, 1, 198.
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265.
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488.
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1α stability in aerobic conditions. Cell Metab. 2016, 24, 740–752.
- Sun, C.; Zhao, Y.; Shi, J.; Zhang, J.; Yuan, Y.; Gu, Y.; Zhang, F.; Gao, X.; Wang, C.; Wang, Y.; et al. Isocitrate dehydrogenase1 mutation reduces the pericyte coverage of microvessels in astrocytic tumours. J. Neuro-Oncol. 2019, 143, 187–196.
- Holroyd, A.K.; Michie, A.M. The role of mTOR-mediated signaling in the regulation of cellular migration. Immunol. Lett. 2018, 196, 74–79.
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018, 9230479.
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-Oncology 2010, 12, 882–889.
- Shahcheraghi, S.H.; Tchokonte-Nana, V.; Lotfi, M.; Lotfi, M.; Ghorbani, A.; Sadeghnia, H.R. Wnt/beta-catenin and PI3K/Akt/mtor Signaling Pathways in Glioblastoma: Two main targets for drug design: A Review. Curr. Pharm. Des. 2020, 26, 1729–1741.
- Carbonneau, M.; Gagné, L.M.; Lalonde, M.-E.; Germain, M.-A.; Motorina, A.; Guiot, M.-C.; Secco, B.; Vincent, E.E.; Tumber, A.; Hulea, L.; et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 2016, 7, 12700.
- Caron, A.; Baraboi, E.D.; Laplante, M.; Richard, D. DEP domain-containing mTOR-interacting protein in the rat brain: Distribution of expression and potential implication. J. Comp. Neurol. 2015, 523, 93–107.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886.
- Caron, A.; Briscoe, D.M.; Richard, D.; Laplante, M. DEPTOR at the Nexus of Cancer, Metabolism, and Immunity. Physiol. Rev. 2018, 98, 1765–1803.
- Liu, Y.; Lu, Y.; Li, A.; Celiku, O.; Han, S.; Qian, M.; Yang, C. Mtorc2/rac1 pathway predisposes cancer aggressiveness in idh1-mutated glioma. Cancers 2020, 12, 787.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
08 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No