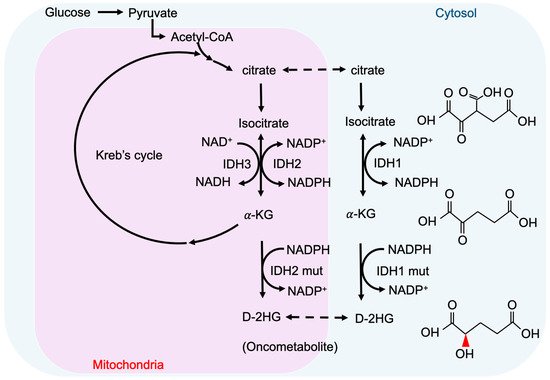
2-hydroxyglutarate (2-HG) is a metabolite detected in urine that was first described by Karl Heinrich Ritthausen in 1868 [
18]. In 1980, Chalmers and Duran identified two similar neurometabolic disorder types related to 2-HG, L-2-hydroxyglutaric aciduria (L-2-HGA) [
19] and D-2-hydroxyglutaric aciduria (D-2-HGA) [
20]. Mutations in L-2-hydroxyglutarate dehydrogenase and D-2-hydroxyglutarate dehydrogenase (D2HGDH) result in the manifestations of L-2-HGA and D-2-HGA, respectively [
21]. Mutations in the mitochondrial citrate carrier SLC25A1 cause combined D-2- and L-2-HGA. Interestingly, the study pointed out half of the patients with D-2-HGA lack the D2HGDH mutation but instead carried mutations in IDH2 [
22]. On the other hand, IDH mutations result in the biosynthesis of D-2-HG from α-ketoglutarate. As mentioned above, somatic mutations in IDH have been identified in glioma and other human malignancies through genome-wide mutation analysis [
2,
23]. To date, cancer-associated IDH1/2 mutations are commonly found in acute myeloid leukemia (~20%) [
24], melanoma [
25], cartilaginous tumors (56–70%) [
26], cholangiocarcinoma (8.5–20%) [
27], and WHO II/III gliomas (~80%) [
3,
28]. There are three IDH isoforms in mammalian cells: one cytosolic form (IDH1) and two mitochondrial forms (IDH2 and IDH3). IDH1 and IDH2 are homodimers, which consume nicotinamide adenine dinucleotide phosphate (NADP
+) for their catalytic function. IDH3 is a heterotetramer and is a nicotinamide adenine dinucleotide (NAD
+)-dependent enzyme. IDH1/2 functions as β-decarboxylating dehydrogenases, which can reversibly convert isocitrate to α-ketoglutarate (α-KG), an essential metabolic intermediate in the Krebs cycle that regulates metabolic and catalytic processes [
29]. Heterozygous IDH1/2 mutations frequently occur in the arginine residues of the catalytic pockets IDH1 (R132H) and IDH2 (R140Q, R172K) [
30,
31]. These IDH1/2 mutations alter the organization of the catalytic centers in these enzymes, which establish gain-of-function changes in their catalytic function, as well as the production of D-2-HG (
Figure 1) [
30,
32,
33,
34]. The chemical structure of D-2-HG is similar to α-KG. The only difference is the carbonyl group in the C2 position that is replaced by the hydroxyl group [
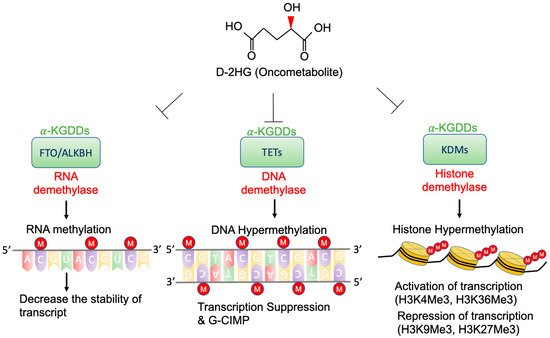
35]. Therefore, D-2-HG could interfere with the enzymes that employ α-KG as a substrate, and competitively inhibit α-KGDDs by occupying the α-KG binding sites in the enzyme [
36]. Moreover, α-KGDDs are a highly diversified enzyme family that is involved in many critical biological processes, such as DNA/histone demethylation, ubiquitination, and hydroxylation, and regulate epigenetic alternation, protein stability, and different signaling (e.g., HIF-1 and mTOR) [
37,
38].
Identifying IDH mutants and their subtypes is a common strategy for molecular pathology in glioma diagnosis. It mainly relies on immunohistochemistry, DNA sequencing, and measurements of intratumoral and circulating D-2-HG, which rely on mass spectrometry (MS)-based [
39,
40], enzymatic assay-based, and magnetic resonance (MR)-based methods [
41]. To precisely detect the levels of D-2-HG in gliomas with an MS-based platform, several studies suggested collecting samples from patients’ cerebrospinal fluid instead of from serum, as CSF has higher D-2-HG concentrations and provides more specific results [
42]. Furthermore, the MS-based method could not clearly distinguish L- and D-2HG, which requires the use of additional chiral derivatization to separate these enantiomers. Although the MS and assay-based methods provide relatively high sensitivity (~ μM), circulating samples must be collected invasively and the D-2-HG final concentration cannot directly reflect the actual tumor size and border. Several recent studies discovered that non-invasive diagnostic approaches, such as magnetic resonance (MR)-based imaging (MRI) [
43] and spectroscopy (MRS) [
44] could be used to predict IDH mutations by measuring D-2-HG in gliomas with mM level sensitivity. Magnetic resonance spectroscopic imaging (MRSI) integrates the information of MRS and MRI, which can detect and quantify various metabolites, and generate the metabolite map from multiple lesions within the brain [
45], which adds value to conventional MRI in pre-operation planning and post-treatment monitoring. MRS-based methods provide 88.6% accuracy in identifying the IDH mutational status, with 89.5% sensitivity and 81.3% specificity, suggesting that MR-based techniques are safe and promising approaches to support glioma diagnosis [
46].