 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stéphane Chavanas | + 2927 word(s) | 2927 | 2021-08-26 04:35:59 | | | |
| 2 | Lindsay Dong | Meta information modification | 2927 | 2021-09-28 08:07:10 | | |
Video Upload Options
Peroxisome Proliferator-Activated Receptor gamma (PPARγ) is a master regulator of metabolism, adipogenesis, inflammation and cell cycle, and it has been extensively studied in the brain in relation to inflammation or neurodegeneration. Specific to viral infections is the ability to subvert signaling pathways of the host cell to ensure virus replication and spreading, as deleterious as the consequences may be for the host.
1. Introduction
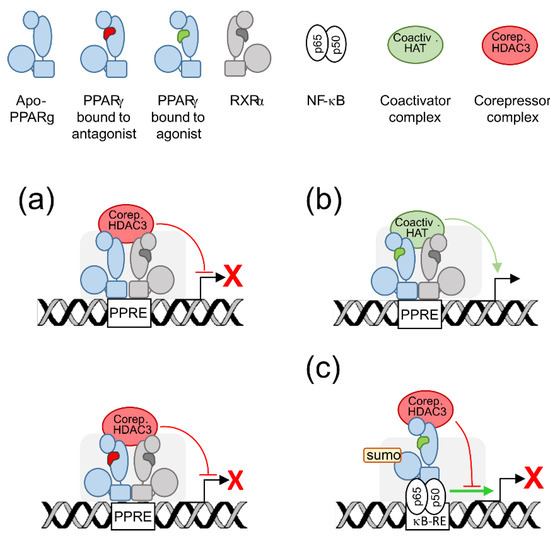
2. PPARγ Molecular Levers
3. PPARγ Expression in the Brain
In a founder study, in situ hybridization analyses of embryonic rat brains revealed transient PPARγ mRNA expression in forebrain, midbrain and, at higher levels, hindbrain, from E13.5, to before E18.5 [3].
Few data are available on PPARγ expression in human brain due to its limited accessibility. We explored PPARγ expression by immunohistological analysis using fetal brain slices from elective abortion [16]. The cases were 23 to 28 gestational weeks and presented with conditions non related to brain such as (1) Digeorges syndrome (ie cardiopathy, endocrinopathy, facial dysplasia), (2) chorioamniotitis and anamnios (i.e., loss of amniotic liquid due to inflammation and premature rupture of membranes), (3) renal failure and (4) atrioventricular canal (heart dysplasia) and omphalocele (defective development of the abdominal wall). In any cases, no PPARγ was detected in any area of the brain parenchyma whereas it was detected in brain blood vessel cells. Soon after, immunofluorescence analysis of superior frontal gyrus (a part of the frontal cortex) from postmortem adult human brain has shown PPARγ expression in neurons and astrocytes but not in microglia [17]. Together those studies underscore that PPARγ is not evenly expressed in the brain, nor is it expressed in the same way in the fetal or adult brain, which raises the possibility that it exerts specific functions apart from its anti-inflammatory and metabolic functions.
4. PPARγ Responds to Specific Issues of the Brain Cell
4.1. Energy Supply, Oxidative Stress, and Mitochondria
PPARγ and/or PPARγ agonists were shown to exert antioxidant functions by upregulating the antioxidant enzymes haem oxygenase-1 (HO-1), catalase or copper/zinc superoxide dismutase (SOD) and downregulating the pro-oxydative enzymes inducible nitric oxide synthase (iNOS) or cyclooxygenase 2 (COX2) (reviewed in [5][7]). Rosiglitazone was also shown to prevent apoptosis related to amyloid [18] or tumor necrosis factor alpha (TNF-α) [19] in human neural stem cells by normalization of oxidative stress and mitochondrial function. Indeed, PPARγ protective role is further supported by its positive effect on mitochondria, that, beyond the cell powerhouse, are key regulators of redox balance [20]. A wealth of in vitro studies reviewed in [5][7] have shown that PPARγ and/or its agonists improved mitochondrial functions in human lymphocytes, adipocytes, astrocytes, neuroblastoma (SH-SY5Y) or neuronal (NT2) cell lines and hippocampal neurons, as shown by increased mitochondrial membrane potential (ΔΨm), increased mitochondrial DNA (mtDNA) copy number, modulation of mitochondrial fusion-fission events and/or expression of factors beneficial to mitochondrial biogenesis and homeostasis, namely the co-activator PGC1-α [21], the mitochondrial transcription factor A (TFAM) [22] or the nuclear factor erythroid-derived 2-like 2 Nrf2 [23].
4.2. Neuroinflammation
Recent findings have provided better knowledge on the protective role of PPARγ in neuroinflammation. PPARγ has been shown to mediate suppression of inflammation by the anesthetic propofol in rat astrocytes [24]. To note, this effect is associated with PPARγ-dependent inhibition of the Wnt/β-catenin pathway, an important pathway which enhances neuroinflammation and has a mutual positive regulation with NF-ĸB [25]. It has been shown that translocator protein (TSPO) inhibited microglia activation by interleukin (IL-) 4 through PPARγ activity in a primary microglia polarization model [26]. Rice bran extract (which is rich in PUFA) as well as pioglitazone have been reported to protect against inflammation induced by lipopolysaccharides (LPS) in a mouse model, decreasing TNF-a and COX2 levels in brain, reducing striatal plaque formation and suppressing cortical and hippocampal tissue damage, all effects requiring PPARγ activity [27]. Other recent studies converged to support positive, PPARγ-dependent, role against neuroinflammation of PPARγ agonists as rosiglitazone which induced IL-10 in primary rat astrocytes exposed to LPS [28], or pioglitazone in a rat model of chronic intermittent hypoxia [29].
4.3. Neurogenesis
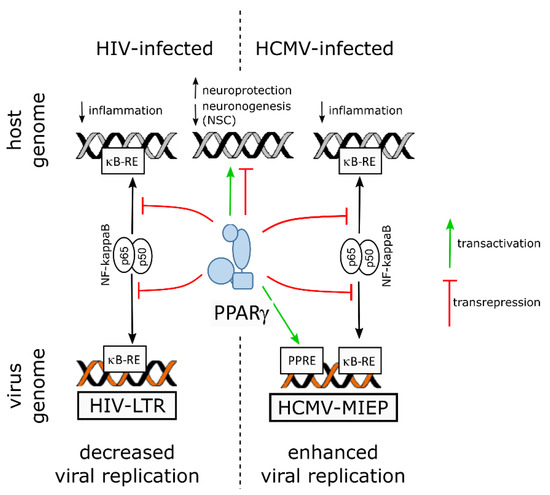
5. PPARγ in the Infected Adult or Developing Brain
5.1. PPARγ, the Adult Brain and Human Immunodeficiency Virus 1
More recent studies have converged to highlight the beneficial role of PPARγ activation in HIV-infected brain. It has been disclosed that insulin treatment upregulated PPARγ expression in HIV-infected primary cultures of human microglia as well as in the cortex, but not in the striatum, of cats infected with feline immunodefiency virus, along with antiviral, anti-inflammatory, and neuroprotective outcomes [33]. Rosiglitazone was found to inhibit NF-κB as well as the release of inflammatory mediators (TNFα, IL-1β) or of iNOS and to prevent downregulation of the mouse ortholog of the glutamate transporter EAAT2 (excitatory amino acid transporter 2) caused by recombinant gp120 in primary mixed cultures of rat astrocytes and microglia or in rat after intracranial injection [34]. Interestingly, the same study reported a decrease in PPARγ transcript levels associated with gp120 treatment. EcoHIV is a chimeric HIV harboring gp80 from murine leukemia virus in place of gp120, thereby allowing for the infection of mouse cells and the onset of some molecular change observed in HAND [35]. Rosiglitazone and pioglitazone were demonstrated to reverse the increase in inflammatory mediators (TNFα, IL-1β, the chemokines CCL2, CCL3, CXCL10) and iNOS levels induced by EcoHIV in primary cultures of mouse glial cells and in mouse brains after intracranial injection [36]. In the same study, the two thiazolidinediones were also found to reduce in vivo EcoHIV p24 protein levels in the brain, what strongly supported an antiviral activity of the two agonists. Since then, similar results were obtained by the same group with the novel, non-thiazolidinedione, PPARγ agonist, INT131 [37]. PPARγ activity was however not assessed in these three reports.
Another role of PPARγ apart from neuroinflammatory modulation, has been highlighted in the context of HIV infection. Blood-brain barrier (BBB) is critical for HIV entry into the brain, and tight junction proteins are key structural and functional elements of integrity and efficiency of the BBB. In an in vitro BBB model, loss of barrier efficiency caused by HIV-infected human monocytes was shown to be reduced by overexpression of PPARγ in monocytes, in particular through repression of HIV-induced matrix metalloproteases (MMP) -2 and -9 activities [38].
On the virus side, NF-ĸB activity is known to be subverted to stimulate viral replication in the host cell by using the two NF-ĸB responsive elements within the promoter enhancer region of the long terminal repeat sequence (LTR) of the HIV genome [10]. Hence, by counteracting NF-kB through transrepression, PPARγ hampers not only inflammatory mediators release but also viral replication. Indeed, PPARγ activity was shown to suppress HIV LTR promoter activity, to decrease NF-κB occupancy of the LTR in infected cell, and, finally, to impair HIV replication in brain macrophages of an humanized mouse model of HIV encephalititis [39].
5.2. PPARγ, the Developing Brain and Zika Virus
Notably, PPARγ transcript levels were found to be increased in human NPCs derived from induced pluripotent stem cells (iPSC), as revealed by RNA-seq, along with productive infection, proliferation arrest and apoptosis ([40], and supplemental data therein). A more recent study used quantitative proteomics and transcriptomics in ZIKV-infected human NPCs and revealed, however, decreased levels of PPARγ mRNA [41]. The same study reported upregulation of RXRγ, of a positive regulator of PPARγ activity (Signal transducer and activator of transcription [STAT] 5 [42]) and of two negative regulators of PPARγ activity (FGR, a member of the Src family of tyrosine protein kinases [43], and the AP-1 transcription factor c-Jun [44]), whereas nuclear receptor coactivator 1 (NCOA1), a coactivator of both RXR and PPARγ [45], was found to be downregulated.
5.3. PPARγ, the Developing Brain and Human Cytomegalovirus

6. Conclusions
References
- Zhu, Y.; Alvares, K.; Huang, Q.; Rao, M.S.; Reddy, J.K. Cloning of a New Member of the Peroxisome Proliferator-Activated Receptor Gene Family from Mouse Liver. J. Biol. Chem. 1993, 268, 26817–26820.
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112.
- Braissant, O.; Wahli, W. Differential Expression of Peroxisome Proliferator-Activated Receptor-α, -Î2, and -Î3 during Rat Embryonic Development. Endocrinology 1998, 139, 2748–2754.
- Wada, K.; Nakajima, A.; Katayama, K.; Kudo, C.; Shibuya, A.; Kubota, N.; Terauchi, Y.; Tachibana, M.; Miyoshi, H.; Kamisaki, Y.; et al. Peroxisome Proliferator-Activated Receptor Gamma-Mediated Regulation of Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem. 2006, 281, 12673–12681.
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2018, 38, 121–132.
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome Proliferator-Activated Receptor γ (PPARγ): A Master Gatekeeper in CNS Injury and Repair. Prog. Neurobiol. 2018, 163-164, 27–58.
- Corona, J.C.; Duchen, M.R. PPARγ as a Therapeutic Target to Rescue Mitochondrial Function in Neurological Disease. Free Radic. Biol. Med. 2016, 100, 153–163.
- Tufano, M.; Pinna, G. Is There a Future for PPARs in the Treatment of Neuropsychiatric Disorders? Molecules 2020, 25, 1062.
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055.
- Hiscott, J.; Kwon, H.; Génin, P. Hostile Takeovers: Viral Appropriation of the NF-ΚB Pathway. J. Clin. Investig. 2001, 107, 143–151.
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The Nuclear Receptor Superfamily: A Structural Perspective. Protein Sci. 2018, 27, 1876–1892.
- Shang, J.; Mosure, S.A.; Zheng, J.; Brust, R.; Bass, J.; Nichols, A.; Solt, L.A.; Griffin, P.R.; Kojetin, D.J. A Molecular Switch Regulating Transcriptional Repression and Activation of PPARγ. Nat. Commun. 2020, 11.
- Tontonoz, P.; Graves, R.A.; Budavari, A.I.; Erdjument-bromage, H.; Lui, M.; Hu, E.; Tempst, P.; Spiegelman, B.M. Adipocyte-Specific Transcription Factor ARF6 Is a Heterodimeric Complex of Two Nuclear Hormone Receptors, PPAR7 and RXRa. Nucleic Acids Res. 1994, 22, 5628–5634.
- Lemay, D.G.; Hwang, D.H. Genome-Wide Identification of Peroxisome Proliferator Response Elements Using Integrated Computational Genomics. J. Lipid Res. 2006, 47, 1583–1587.
- Wahli, W. A Gut Feeling of the PXR, PPAR and NF-kappaB Connection. J. Int. Med. 2008, 263, 613–619.
- Rolland, M.; Li, X.; Sellier, Y.; Martin, H.; Perez-Berezo, T.; Rauwel, B.; Benchoua, A.; Bessières, B.; Aziza, J.; Cenac, N.; et al. PPARγ Is Activated during Congenital Cytomegalovirus Infection and Inhibits Neuronogenesis from Human Neural Stem Cells. PLoS Pathog. 2016, 12, e1005547.
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6, 27618.
- Chiang, M.C.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Yen, C.H.; Lin, C.H. Rosiglitazone Activation of PPARγ-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Mitochondrial Dysfunction and Oxidative Stress. Neurobiol. Aging 2016, 40, 181–190.
- Chiang, M.C.; Cheng, Y.C.; Lin, K.H.; Yen, C.H. PPARγ Regulates the Mitochondrial Dysfunction in Human Neural Stem Cells with Tumor Necrosis Factor Alpha. Neuroscience 2013, 229, 118–129.
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607.
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1269–1278.
- Kang, I.; Chu, C.T.; Kaufman, B.A. The Mitochondrial Transcription Factor TFAM in Neurodegeneration: Emerging Evidence and Mechanisms. FEBS Lett. 2018, 592, 793–811.
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617.
- Jiang, P.; Jiang, Q.; Yan, Y.; Hou, Z.; Luo, D. Propofol Ameliorates Neuropathic Pain and Neuroinflammation through PPAR γ Up-Regulation to Block Wnt/β-Catenin Pathway. Neurol. Res. 2021, 43, 71–77.
- Vallée, A.; Vallée, J.N.; Guillevin, R.; Lecarpentier, Y. Interactions Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma on Neuroinflammation, Demyelination, and Remyelination in Multiple Sclerosis. Cell. Mol. Neurobiol. 2018, 38, 783–795.
- Zhou, D.; Ji, L.; Chen, Y. TSPO Modulates IL-4-Induced Microglia/Macrophage M2 Polarization via PPAR-γ Pathway. J. Mol. Neurosci. 2020, 70, 542–549.
- Abd El Fattah, M.A.; Abdelhamid, Y.A.; Elyamany, M.F.; Badary, O.A.; Heikal, O.A. Rice Bran Extract Protected against LPS-Induced Neuroinflammation in Mice through Targeting PPAR-γ Nuclear Receptor. Mol. Neurobiol. 2021, 58, 1504–1516.
- Chistyakov, D.V.; Astakhova, A.A.; Goriainov, S.V.; Sergeeva, M.G. Comparison of PPAR Ligands as Modulators of Resolution of Inflammation, via Their Influence on Cytokines and Oxylipins Release in Astrocytes. Int. J. Mol. Sci. 2020, 21, 9577.
- Zhang, X.; Li, N.; Lu, L.; Lin, Q.; Li, L.; Dong, P.; Yang, B.; Li, D.; Fei, J. Pioglitazone Prevents Sevoflurane-induced Neuroinflammation and Cognitive Decline in a Rat Model of Chronic Intermittent Hypoxia by Upregulating Hippocampal PPAR-γ. Mol. Med. Rep. 2019, 49, 3815–3822.
- Stergiopoulos, A.; Politis, P.K. The Role of Nuclear Receptors in Controlling the Fine Balance between Proliferation and Differentiation of Neural Stem Cells. Arch. Biochem. Biophys. 2013, 534, 27–37.
- Gkikas, D.; Tsampoula, M.; Politis, P.K. Nuclear Receptors in Neural Stem/Progenitor Cell Homeostasis. Cell. Mol. Life Sci. 2017, 74, 4097–4120.
- De Nuccio, C.; Bernardo, A.; Troiano, C.; Brignone, M.S.; Falchi, M.; Greco, A.; Rosini, M.; Basagni, F.; Lanni, C.; Serafini, M.M.; et al. Nrf2 and Ppar-γ Pathways in Oligodendrocyte Progenitors: Focus on Ros Protection, Mitochondrial Biogenesis and Promotion of Cell Differentiation. Int. J. Mol. Sci. 2020, 21, 7216.
- Mamik, M.K.; Asahchop, E.L.; Chan, W.F.; Zhu, Y.; Branton, W.G.; McKenzie, B.A.; Cohen, E.A.; Power, C. Insulin Treatment Prevents Neuroinflammation and Neuronal Injury with Restored Neurobehavioral Function in Models of HIV/AIDS Neurodegeneration. J. Neurosci. 2016, 36, 1683–1695.
- Omeragic, A.; Hoque, M.T.; Choi, U.; Bendayan, R. Peroxisome Proliferator-Activated Receptor-Gamma: Potential Molecular Therapeutic Target for HIV-1-Associated Brain Inflammation. J. Neuroinflamm. 2017, 14.
- Potash, M.J.; Chao, W.; Bentsman, G.; Paris, N.; Saini, M.; Nitkiewicz, J.; Belem, P.; Sharer, L.; Brooks, A.I.; Volsky, D.J. A Mouse Model for Study of Systemic HIV-1 Infection, Antiviral Immune Responses, and Neuroinvasiveness. Proc. Natl. Acad. Sci. USA 2005, 102, 3760–3765.
- Omeragic, A.; Kara-Yacoubian, N.; Kelschenbach, J.; Sahin, C.; Cummins, C.L.; Volsky, D.J.; Bendayan, R. Peroxisome Proliferator-Activated Receptor-Gamma Agonists Exhibit Anti-Inflammatory and Antiviral Effects in an EcoHIV Mouse Model. Sci. Rep. 2019, 9.
- Omeragic, A.; Saikali, M.F.; Currier, S.; Volsky, D.J.; Cummins, C.L.; Bendayan, R. Selective Peroxisome Proliferator-Activated Receptor-Gamma Modulator, INT131 Exhibits Anti-Inflammatory Effects in an EcoHIV Mouse Model. FASEB J. 2020, 34, 1996–2010.
- Huang, W.; Eum, S.Y.; András, I.E.; Hennig, B.; Toborek, M. PPARα and PPARγ Attenuate HIV-induced Dysregulation of Tight Junction Proteins by Modulations of Matrix Metalloproteinase and Proteasome Activities. FASEB J. 2009, 23, 1596–1606.
- Potula, R.; Ramirez, S.H.; Knipe, B.; Leibhart, J.; Schall, K.; Heilman, D.; Morsey, B.; Mercer, A.; Papugani, A.; Dou, H.; et al. Peroxisome Proliferator-Activated Receptor-γ Activation Suppresses HIV-1 Replication in an Animal Model of Encephalitis. AIDS 2008, 22, 1539–1549.
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590.
- Thulasi Raman, S.N.; Latreille, E.; Gao, J.; Zhang, W.; Wu, J.; Russell, M.S.; Walrond, L.; Cyr, T.; Lavoie, J.R.; Safronetz, D.; et al. Dysregulation of Ephrin Receptor and PPAR Signaling Pathways in Neural Progenitor Cells Infected by Zika Virus. Emerg. Microbes Infect. 2020, 9, 2046–2060.
- Sharma, R.; Luong, Q.; Sharma, V.M.; Harberson, M.; Harper, B.; Colborn, A.; Berryman, D.E.; Jessen, N.; Jørgensen, J.O.L.; Kopchick, J.J.; et al. Growth Hormone Controls Lipolysis by Regulation of FSP27 Expression. J. Endocrinol. 2018, 239, 289–301.
- Hua, T.N.M.; Kim, M.K.; Vo, V.T.A.; Choi, J.W.; Choi, J.H.; Kim, H.W.; Cha, S.K.; Park, K.S.; Jeong, Y. Inhibition of Oncogenic Src Induces FABP4-Mediated Lipolysis via PPARγ Activation Exerting Cancer Growth Suppression. EBioMedicine 2019, 41, 134–145.
- Ban, K.; Peng, Z.; Lin, W.; Kozar, R.A. Arginine Decreases Peroxisome Proliferator-Activated Receptor-γ Activity via c-Jun. Mol. Cell. Biochem. 2012, 362, 7–13.
- Triki, M.; Lapierre, M.; Cavailles, V.; Mokdad-Gargouri, R. Expression and Role of Nuclear Receptor Coregulators in Colorectal Cancer. World J. Gastroenterol. 2017, 23, 4480.
- Han, D.; Byun, S.-H.; Kim, J.; Kwon, M.; Pleasure, S.J.; Ahn, J.-H.; Yoon, K. Human Cytomegalovirus IE2 Protein Disturbs Brain Development by the Dysregulation of Neural Stem Cell Maintenance and the Polarization of Migrating Neurons. J. Virol. 2017, 91.
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, A.; Söderberg-Nauclér, C. Human Cytomegalovirus Inhibits Neuronal Differentiation and Induces Apoptosis in Human Neural Precursor Cells. J. Virol. 2006, 80, 8929–8939.
- Luo, M.H.; Hannemann, H.; Kulkarni, A.S.; Schwartz, P.H.; O’Dowd, J.M.; Fortunato, E.A. Human Cytomegalovirus Infection Causes Premature and Abnormal Differentiation of Human Neural Progenitor Cells. J. Virol. 2010, 84, 3528–3541.
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human Cytomegalovirus Infection of Human Embryonic Stem Cell-Derived Primitive Neural Stem Cells Is Restricted at Several Steps but Leads to the Persistence of Viral DNA. J. Virol. 2014, 88, 4021–4039.
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Sundtröm, E.; Seiger, Å.; Söderberg-Nauclér, C. Late Human Cytomegalovirus (HCMV) Proteins Inhibit Differentiation of Human Neural Precursor Cells into Astrocytes. J. Neurosci. Res. 2007, 85, 583–593.
- D’Aiuto, L.; Di Maio, R.; Heath, B.; Raimondi, G.; Milosevic, J.; Watson, A.M.; Bamne, M.; Parks, W.T.; Yang, L.; Lin, B.; et al. Human Induced Pluripotent Stem Cell-Derived Models to Investigate Human Cytomegalovirus Infection in Neural Cells. PLoS ONE 2012, 7, e49700.
- Rolland, M.; Martin, H.; Bergamelli, M.; Sellier, Y.; Bessières, B.; Aziza, J.; Benchoua, A.; Leruez-Ville, M.; Gonzalez-Dunia, D.; Chavanas, S. Human Cytomegalovirus Infection Is Associated with Increased Expression of the Lissencephaly Gene PAFAH1B1 Encoding LIS1 in Neural Stem Cells and Congenitally Infected Brains. J. Pathol. 2021, 254, 92–102.
- Leghmar, K.; Cenac, N.; Rolland, M.; Martin, H.; Rauwel, B.; Bertrand-Michel, J.; Le Faouder, P.; Bénard, M.; Casper, C.; Davrinche, C.; et al. Cytomegalovirus Infection Triggers the Secretion of the PPARgamma Agonists 15-Hydroxyeicosatetraenoic Acid (15-HETE) and 13-Hydroxyoctadecadienoic Acid (13-HODE) in Human Cytotrophoblasts and Placental Cultures. PLoS ONE 2015, 10, e0132627.
- Stump, M.; Guo, D.F.; Lu, K.T.; Mukohda, M.; Cassell, M.D.; Norris, A.W.; Rahmouni, K.; Sigmund, C.D. Nervous System Expression of PPARγ and Mutant PPARγ Has Profound Effects on Metabolic Regulation and Brain Development. Endocrinology 2016, 157, 4266–4275.
- Kamin, D.; Hadigan, C.; Lehrke, M.; Mazza, S.; Lazar, M.A.; Grinspoon, S. Resistin Levels in Human Immunodeficiency Virus-Infected Patients with Lipoatrophy Decrease in Response to Rosiglitazone. J. Clin. Endocrinol. Metab. 2005, 90, 3423–3426.


