Peroxisome Proliferator-Activated Receptor gamma (PPARγ) is a master regulator of metabolism, adipogenesis, inflammation and cell cycle, and it has been extensively studied in the brain in relation to inflammation or neurodegeneration. Specific to viral infections is the ability to subvert signaling pathways of the host cell to ensure virus replication and spreading, as deleterious as the consequences may be for the host.
1. Introduction
Peroxisome Proliferator-Activated Receptor gamma (PPARγ) was discovered and cloned almost 30 years ago, as a new member of a family of receptors activated in response to treatment of liver cells by an heterogeneous group of chemicals, namely peroxysome proliferators [
1]. Since then, an ever growing body of research has provided us with a better knowledge about PPARγ, which is now known as a master regulator of gene expression in lipid and glucose metabolism, adipogenesis, inflammation, cell proliferation and cancer [
2].
It has been almost 25 years since PPARγ transcripts were detected in brain of rat embryos [
3]. This early finding suggested that PPARγ might be of importance in brain development; an assumption that was strengthened thereafter by the observation of a «disorganized brain» in
Pparg knock-out mouse embryos [
4]. PPARγ in the brain has been extensively studied in relation to inflammation or neurodegeneration [
5]. A wealth of in vitro, in vivo and clinical studies have shown that PPARγ plays a beneficial role on brain injury [
6] and neurodegenerative disorders such as Multiple Sclerosis, Alzheimer’s disease and Amyotrophic Lateral Sclerosis [
7]. Also, on the bases of encouraging preclinical studies, PPARγ has been proposed as a possible therapeutic target for psychiatric disorders [
8] or drug addiction and substance abuse [
9]. Although the role of PPARγ in the regulation of the immune response and inflammation is well established, little is known however about its role in infections of the brain parenchyma, particularly viral infections.
A better understanding on the role of PPARγ in the infected brain may help designing new therapeutic strategies. Furthermore, specific to viral infections is the ability to subvert signaling pathways of the host cell in order to ensure viral replication and spread, as deleterious as the consequences may be for the host. For example, many viruses have evolved mechanisms to regulate positively or negatively activity of the nuclear factor ĸB (NF-ĸB) to facilitate their replication, host cell survival, or immuno-evasion [
13]. In this respect, the pleiotropic role of PPARγ makes it an expected critical target of infection. Thus, investigating PPARγ in neural cell infections can provide insight on the molecular and cellular outcomes of PPARγ activity in the healthy cell as well as the infected cell.
2. PPARγ Molecular Levers
Peroxysome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily [
14]. As such, they are activated by lipophilic, membrane-permeant, ligands. Upon ligand binding, nuclear receptors form homo- or hetero-dimers and translocate to the nucleus to regulate gene transcription. PPAR family comprises three members, PPARα, PPARβ/δ, and PPARγ. They share a common structure containing six highly conserved functional domains: a first transcription activation function domain (AF-1), a two zinc-fingers DNA binding domain (DBD), a hinge domain, a ligand binding domain (LBD) and a second activation function domain (AF-2) that modulates binding to either co-activator or repressor factors in a ligand-dependent fashion [
14,
15]. The gene encoding PPARγ, namely
PPARG, has a complex pattern of expression. Two alternative promoters and alternative splicing events can generate seven
PPARG transcripts translated to two PPARγ isoforms: the widely expressed PPARγ1 and the adipocyte-restricted PPARγ2 [
2].
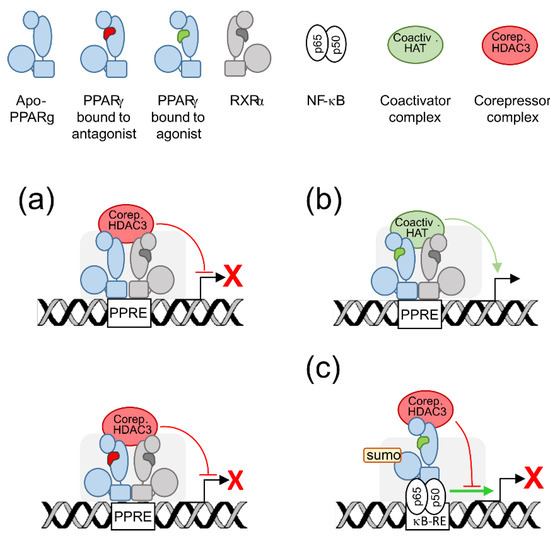
Transactivation and transrepression refer to positive or negative gene transcriptional regulation by PPARγ, respectively. Transactivation requires both DNA-binding and agonist-binding whereas transrepression may require or not DNA-binding (Figure 1).
Figure 1. Graphical summary of PPARγ transactivating and transrepressing activities. (a) DNA binding-dependent transrepression: unbound PPARγ (top) or PPARγ bound to an antagonist (bottom) forms a dimer with RXRα, binds to a cognate response element (PPRE) and recruits corepressors (Corep.) and HDAC3 to assemble a repressive complex which blocks transcription of the cognate gene (broken arrow). (b) Transactivation: agonist-bound PPARγ and RXRα, bound to a PPRE, recruit coactivators (Coactiv.) and HAT to assemble a permissive complex which enhances transcription of the cognate gene. (c) DNA binding-independent transrepression: ligand-activated PPARγ and corepressor complex bind to a target transcription factor as NF-ĸB to prevent it from activating cognate gene transcription. Sumoylation (sumo) increases the stability of the complex PPARγ-corepressor. ĸB-RE: NF-ĸB response element.
PPARγ forms dimers with another nuclear receptor, namely the retinoid X receptor alpha (RXRα) whose ligand is 9-cis retinoic acid [
16]. The PPARγ-RXRα dimer translocates to the nucleus and binds cognate DNA sequences named PPAR Responsive Elements (PPRE) [
17]. For transactivation, the dimer formed by agonist-bound PPARγ and RXRα recruits coactivators such as PPARγ coactivator 1-α (PGC-1α), E1A binding protein p300 (EP300), or steroid receptor coactivator (SRC1), and histone acetyl transferases (HAT) to assemble a permissive complex on target gene promoters or enhancers, what results in focal chromatin relaxation and enhanced transcription of the cognate gene [
2] (
Figure 1). This is how PPARγ transactivates expression of a wealth of neuroprotective genes critical for mitochondria, microglial regulation and oxidative stress management [
2]. PPARγ also exerts DNA-binding independent transrepression. When activated by an agonist, PPARγ bound to corepressors can bind to other transcription factors such as nuclear factor ĸB (NF-ĸB) or activating protein 1 (AP-1) to prevent them from activating inflammatory gene transcription [
2] (
Figure 1). The PPARγ-corepressor complex can also promote NF-ĸB degradation or export out of the nucleus [
6,
18]. These transrepressive mecanisms underlie the anti-inflammatory action of PPARγ [
2].
3. PPARγ Expression in the Brain
In a founder study, in situ hybridization analyses of embryonic rat brains revealed transient PPARγ mRNA expression in forebrain, midbrain and, at higher levels, hindbrain, from E13.5, to before E18.5 [3].
Few data are available on PPARγ expression in human brain due to its limited accessibility. We explored PPARγ expression by immunohistological analysis using fetal brain slices from elective abortion [31]. The cases were 23 to 28 gestational weeks and presented with conditions non related to brain such as (1) Digeorges syndrome (ie cardiopathy, endocrinopathy, facial dysplasia), (2) chorioamniotitis and anamnios (i.e., loss of amniotic liquid due to inflammation and premature rupture of membranes), (3) renal failure and (4) atrioventricular canal (heart dysplasia) and omphalocele (defective development of the abdominal wall). In any cases, no PPARγ was detected in any area of the brain parenchyma whereas it was detected in brain blood vessel cells. Soon after, immunofluorescence analysis of superior frontal gyrus (a part of the frontal cortex) from postmortem adult human brain has shown PPARγ expression in neurons and astrocytes but not in microglia [37]. Together those studies underscore that PPARγ is not evenly expressed in the brain, nor is it expressed in the same way in the fetal or adult brain, which raises the possibility that it exerts specific functions apart from its anti-inflammatory and metabolic functions.
4. PPARγ Responds to Specific Issues of the Brain Cell
4.1. Energy Supply, Oxidative Stress, and Mitochondria
PPARγ and/or PPARγ agonists were shown to exert antioxidant functions by upregulating the antioxidant enzymes haem oxygenase-1 (HO-1), catalase or copper/zinc superoxide dismutase (SOD) and downregulating the pro-oxydative enzymes inducible nitric oxide synthase (iNOS) or cyclooxygenase 2 (COX2) (reviewed in [5,7]). Rosiglitazone was also shown to prevent apoptosis related to amyloid [42] or tumor necrosis factor alpha (TNF-α) [43] in human neural stem cells by normalization of oxidative stress and mitochondrial function. Indeed, PPARγ protective role is further supported by its positive effect on mitochondria, that, beyond the cell powerhouse, are key regulators of redox balance [44]. A wealth of in vitro studies reviewed in [5,7] have shown that PPARγ and/or its agonists improved mitochondrial functions in human lymphocytes, adipocytes, astrocytes, neuroblastoma (SH-SY5Y) or neuronal (NT2) cell lines and hippocampal neurons, as shown by increased mitochondrial membrane potential (ΔΨm), increased mitochondrial DNA (mtDNA) copy number, modulation of mitochondrial fusion-fission events and/or expression of factors beneficial to mitochondrial biogenesis and homeostasis, namely the co-activator PGC1-α [45], the mitochondrial transcription factor A (TFAM) [46] or the nuclear factor erythroid-derived 2-like 2 Nrf2 [47].
4.2. Neuroinflammation
Recent findings have provided better knowledge on the protective role of PPARγ in neuroinflammation. PPARγ has been shown to mediate suppression of inflammation by the anesthetic propofol in rat astrocytes [57]. To note, this effect is associated with PPARγ-dependent inhibition of the Wnt/β-catenin pathway, an important pathway which enhances neuroinflammation and has a mutual positive regulation with NF-ĸB [58]. It has been shown that translocator protein (TSPO) inhibited microglia activation by interleukin (IL-) 4 through PPARγ activity in a primary microglia polarization model [59]. Rice bran extract (which is rich in PUFA) as well as pioglitazone have been reported to protect against inflammation induced by lipopolysaccharides (LPS) in a mouse model, decreasing TNF-a and COX2 levels in brain, reducing striatal plaque formation and suppressing cortical and hippocampal tissue damage, all effects requiring PPARγ activity [60]. Other recent studies converged to support positive, PPARγ-dependent, role against neuroinflammation of PPARγ agonists as rosiglitazone which induced IL-10 in primary rat astrocytes exposed to LPS [61], or pioglitazone in a rat model of chronic intermittent hypoxia [62].
4.3. Neurogenesis
In the embryo, PPARγ has been shown to support NPC proliferation, trigger astrogliogenesis, inhibit neuron production (neuronogenesis) and enhance neurite outgrowth of differentiating neurons, whereas in the adult brain, PPARγ has been reported to enhance NSC self-renewal and differentiation [
68]. A wealth of studies recently reviewed in [
69] showed that PPARγ supports NSC growth, survival and stemness maintenance and positively regulates neuronogenesis and neurite outgrowth in maturing neurons. More recently, pioglitazone was shown to promote differentiation of rat primary oligodendrocytes [
49].
5. PPARγ in the Infected Adult or Developing Brain
5.1. PPARγ, the Adult Brain and Human Immunodeficiency Virus 1
More recent studies have converged to highlight the beneficial role of PPARγ activation in HIV-infected brain. It has been disclosed that insulin treatment upregulated PPARγ expression in HIV-infected primary cultures of human microglia as well as in the cortex, but not in the striatum, of cats infected with feline immunodefiency virus, along with antiviral, anti-inflammatory, and neuroprotective outcomes [85]. Rosiglitazone was found to inhibit NF-κB as well as the release of inflammatory mediators (TNFα, IL-1β) or of iNOS and to prevent downregulation of the mouse ortholog of the glutamate transporter EAAT2 (excitatory amino acid transporter 2) caused by recombinant gp120 in primary mixed cultures of rat astrocytes and microglia or in rat after intracranial injection [86]. Interestingly, the same study reported a decrease in PPARγ transcript levels associated with gp120 treatment. EcoHIV is a chimeric HIV harboring gp80 from murine leukemia virus in place of gp120, thereby allowing for the infection of mouse cells and the onset of some molecular change observed in HAND [87]. Rosiglitazone and pioglitazone were demonstrated to reverse the increase in inflammatory mediators (TNFα, IL-1β, the chemokines CCL2, CCL3, CXCL10) and iNOS levels induced by EcoHIV in primary cultures of mouse glial cells and in mouse brains after intracranial injection [88]. In the same study, the two thiazolidinediones were also found to reduce in vivo EcoHIV p24 protein levels in the brain, what strongly supported an antiviral activity of the two agonists. Since then, similar results were obtained by the same group with the novel, non-thiazolidinedione, PPARγ agonist, INT131 [89]. PPARγ activity was however not assessed in these three reports.
Another role of PPARγ apart from neuroinflammatory modulation, has been highlighted in the context of HIV infection. Blood-brain barrier (BBB) is critical for HIV entry into the brain, and tight junction proteins are key structural and functional elements of integrity and efficiency of the BBB. In an in vitro BBB model, loss of barrier efficiency caused by HIV-infected human monocytes was shown to be reduced by overexpression of PPARγ in monocytes, in particular through repression of HIV-induced matrix metalloproteases (MMP) -2 and -9 activities [90].
On the virus side, NF-ĸB activity is known to be subverted to stimulate viral replication in the host cell by using the two NF-ĸB responsive elements within the promoter enhancer region of the long terminal repeat sequence (LTR) of the HIV genome [13]. Hence, by counteracting NF-kB through transrepression, PPARγ hampers not only inflammatory mediators release but also viral replication. Indeed, PPARγ activity was shown to suppress HIV LTR promoter activity, to decrease NF-κB occupancy of the LTR in infected cell, and, finally, to impair HIV replication in brain macrophages of an humanized mouse model of HIV encephalititis [96].
5.2. PPARγ, the Developing Brain and Zika Virus
Notably, PPARγ transcript levels were found to be increased in human NPCs derived from induced pluripotent stem cells (iPSC), as revealed by RNA-seq, along with productive infection, proliferation arrest and apoptosis ([99], and supplemental data therein). A more recent study used quantitative proteomics and transcriptomics in ZIKV-infected human NPCs and revealed, however, decreased levels of PPARγ mRNA [100]. The same study reported upregulation of RXRγ, of a positive regulator of PPARγ activity (Signal transducer and activator of transcription [STAT] 5 [101]) and of two negative regulators of PPARγ activity (FGR, a member of the Src family of tyrosine protein kinases [102], and the AP-1 transcription factor c-Jun [103]), whereas nuclear receptor coactivator 1 (NCOA1), a coactivator of both RXR and PPARγ [104], was found to be downregulated.
5.3. PPARγ, the Developing Brain and Human Cytomegalovirus
Infection of neural progenitor cells in the developing brain is thought to be a primary cause of the neurological sequelae due to HCMV congenital infection ([
31] and references therein). In vitro studies showed that HCMV infection of progenitors disrupted self-renewal and polarization [
107], apoptosis [
108], differentiation [
107,
108,
109,
110,
111,
112] or migratory abilities [
113]. Because PPARγ had been shown previously to be upregulated in human placenta cells infected by HCMV [
114], NSCs from human embryonic stem cells were used as a model to investigate the outcomes on PPARγ activity of the infection of neural progenitors by HCMV (
Figure 2) [
31].
Figure 2. Transmission electron microscopy of human NSC cultures infected by HCMV. (a) Representative HCMV-infected NSC, containing numerous electron-dense lipid droplets (asterisks) consistent with active PPARγ. Scale bar: 5 µm. (b) Representative view of the cytoplasm of an infected NSC, containing morphologically mature viral particles (arrowheads) mitochondria (m) and still lipid droplets. Scale bar: 0.5 µm. (c) View of a HCMV particle shedding from an infected cell (top) and the same view after image processing (bottom) to highlight plasma membrane (red dotted line), the exocytosis cavity (B), the viral capsid (C) containing the electron-dense viral chromatin and the viral envelope (E). Scale bar: 0.2 µm.
Infection by HCMV was found to dramatically impair neuronal differentiation of NSCs [
31]. PPARγ was barely detectable in uninfected NSCs whereas nuclei of infected NSCs showed strong immunoreactivity to PPARγ, indicating increased expression and activity of PPARγ [
31]. This result was confirmed by chromatin immunoprecipitation, reporter gene assay or cellular lipid droplet staining. More importantly, this finding was strongly supported by the immunodetection of nuclear PPARγ specifically in the brain germinative zones of congenitally infected fetuses (N = 20) but not in control samples [
31]. Lipidomic analysis revealed that levels of 9-HODE were significantly and specifically increased in infected NSCs, indicating that 9-HODE was the agonist associated with PPARγ activation. 9-HODE was also found to dramatically increase PPARγ levels and activity in uninfected NSCs, recapitulating the effect of infection [
31]. Furthermore, 9-HODE treatment and/or single-out expression of PPARγ were sufficient to impair neuronogenesis of uninfected NSCs, whereas treatment of HCMV-infected NSCs with the PPARγ antagonist T0070907 restored a normal rate of differentiation [
31]. Together these findings revealed that PPARγ exerts a negative role on NSC differentiation to neurons, should they be infected by HCMV or not. This has been supported soon after in another study which demonstrated that conditionally forced expression of Pparγ in mouse neural progenitors resulted in severe microcephaly and brain malformation [
115].
6. Conclusions
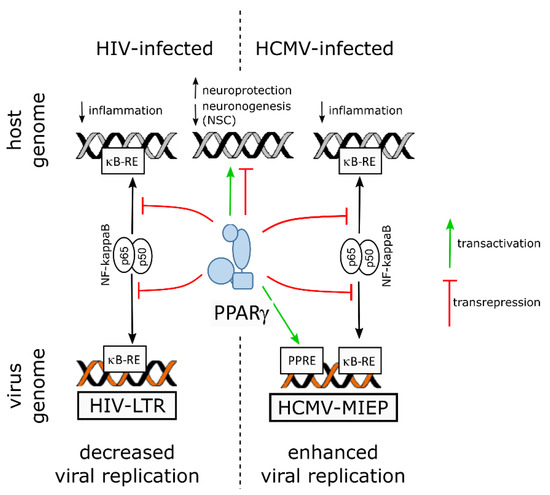
Probably because of its multifaceted role at the crossroads of inflammation, metabolism and cell differentiation, PPARγ can be a double-edged sword in viral infections of neural cells: besides its role in both moderating inflammation and supporting host cell survival, it can be deleterious to neuronal differentiation of progenitors, and either inhibit or support viral replication (Figure 3).
Figure 3. Graphical summary of PPARγ involvement during infection by HIV (left) or HCMV (right). In HIV-infected cells, PPARγ inhibits NF-kB by transrepression (red lines), thereby downregulating inflammatory genes and decreasing the efficiency of viral replication. In contrast, in HCMV-infected cells, PPARγ enhances viral replication by transactivation (green arrow) of the HCMV major immediate early promoter (MIEP) through two PPAR responsive elements (PPRE). In both cases, PPARγ regulates expression of the host cell genome, contributing to neuroprotection and, in neural stem cells (NSC), inhibition of neuronogenesis. ĸB-RE: NF-γB responsive element.
In the infected adult brain, the role of PPARγ in the host response to infection appeared beneficial against inflammation, oxidative stress and viral replication, as exemplified in HIV infection (
Figure 3). PPARγ agonists have been proposed to be promising candidate drugs in the treatment of HIV-1 brain inflammation and neurocognitive outcomes [
86], especially as they are already being used in treatment of HIV-associated lipodystrophy [
121]. In contrast, in the developing brain, PPARγ activation has deleterious outcomes on neurogenesis, as shown in HCMV infection, and possibly in ZIKV infection. Notably, the activation of PPARγ in infection by HCMV is beneficial to viral replication (
Figure 3).
Viruses undergo evolutionary pressure which optimizes both their spreading efficiency and the survival of their host. Whereas both the genomes of HIV and HCMV contain responsive elements to NF-ĸB, HCMV genome has evolved to gain two PPAR responsive elements within its major promoter. These responsive elements allow the subversion of PPARγ activity in the benefit of HCMV replication. Moreover, NF-ĸB transrepression by activated PPARγ accounts for immune evasion.
Yet, it is important to recall how variable the severity of neurological sequelae of HCMV infection may be. Host genetic factors still to be discovered may be important determinants of the severity of the sequelae, as, for example, cis-acting transcriptional regulators of PPARγ gene expression, or reciprocally, putative PPRE within PPARγ target genes.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22168876