 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Uday Ghoshal | + 2273 word(s) | 2273 | 2021-09-01 10:43:59 | | | |
| 2 | Dean Liu | Meta information modification | 2273 | 2021-09-02 05:26:20 | | |
Video Upload Options
The H. pyloriinfection generates an inflammatory reaction in the stomach, resulting in the loss of parietal cells and an elevation in gastric pH. H. pylorimay contribute to microbial dysbiosis, and effective eradication can restore the gut microbiota to a state comparable to that of uninfected people.
1. Introduction
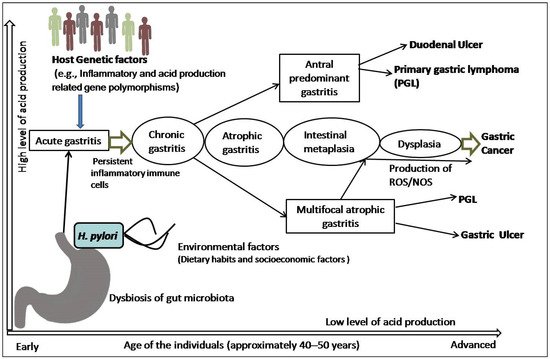
Gastric cancer (GC) is the most common cancer contributing to 5.5% of all new cases of cancers. Moreover, it is the fourth most lethal cancer, resulting in 7.7% of all deaths worldwide [1]. It has a poor prognosis, with a survival rate of less than 5 years among 80% of cases [2]. Although the incidence of GC has decreased, it remains a significant global health burden, with the highest burden in Asia [1]. Helicobacter pylori (H. pylori) infects over half of the world’s population, and this bacterium is the main cause of non-cardia GC [3][4][5]. However, geographical variations of bacterial virulence, age of acquisition of infection, host genetics, and environmental factors may lead to variation in the incidence of GC [6]. Paleo Correa’s hypothesis suggests that gastric carcinogenesis progresses in multiple stages and is caused by various factors. The histological cascade associated with GC is well characterized. It proceeds from normal mucosa infected with H. pylori to chronic active gastritis, atrophic gastritis (AG), intestinal metaplasia (complete at first, then incomplete later), dysplasia, hyperplasia, and adenocarcinoma [7][8][9][10].
A chronic inflammatory state is known to be critical for H. pylori -induced GC; the molecular mechanisms underlying how H. pylori communicate with gastric epithelial cells directly or indirectly to trigger gastric carcinogenesis remain unknown. GC exhibits a multi-factorial etiology. Moreover, owing to some paradoxical observations, it is now speculated that in addition to H. pylori infection, diet and host genetic factors are responsible for the progress from H. pylori -induced inflammation to GC [9][11][12]. A few recent studies also proposed that the increasing prevalence of autoimmune gastritis and dysbiosis of gut microbiota and increased use of antibiotics and acid suppressants, might have led to the variation in the risk of GC, primary gastric lymphoma (PGL), and neuroendocrine carcinoma [13][14][15]. Inter-individual variations on disease susceptibility of GC can also be influenced by other infections such as Epstein-Barr virus, intestinal helminths, and host genetic differences in cytokine genes [9][16][17].
2. Mechanism of Chronic Inflammation and Multi-Step Sequel of Gastric Carcinogenesis
GC is an inflammation-associated carcinoma promoted by H. pylori infection, characterized by ongoing chronic gastritis, formation of metaplastic epithelia, and finally genetic instability in the gastric mucosal epithelium [18][19]. The relationship between chronic inflammation and cancer dates back to Virchow, who, in 1863, hypothesized that the origin of cancer was at the sites of chronic inflammation [20]. Many shreds of evidence proved that the inflammation might result from persistent mucosal or epithelial cell colonization by H. pylori, which may cause GC [11][21][22]. Persistent inflammation leads to increased cellular turnover, especially in the epithelium, and provides selection pressure, which may result in the emergence of cells at high risk for malignant transformation [23]. The association of inflammatory signals with intracellular pathways in gastric epithelial cells eventually leads to uncontrolled cell division, and differentiation remains inadequate. Whereas acute injury and inflammation associated with healing are usually self-limited, chronic injuries or inflammation over decade’s leads to a sustained expansion of proliferative tissue zones that are predisposed to neoplastic progression [24].
The precancerous cascade advances slowly and steadily, and it may take years or several decades to develop malignancy since the initiation of the cascade. However, the rate of progression from metaplasia to dysplasia is not the same in all individuals. A study revealed that the progression rate was two times higher in subjects more than 40 years of age than their younger counterparts [25]. In a subset of patients, this inflammatory process leads to loss of parietal cells and development of AG, followed by intestinal metaplasia (IM), dysplasia, and cancer [26]. Surprisingly, the majority of patients infected with H. pylori not only incline towards the development of pre-malignant lesions or GC, but also towards other gastroduodenal diseases ( Figure 1 ). It is also speculated that all the stages before the development of dysplasia are reversible, although this is still somewhat controversial [24][25][26].
3. Risk Modulation of GC by Co-Existence of Gut Microbiota and Infestation with Intestinal Helminths
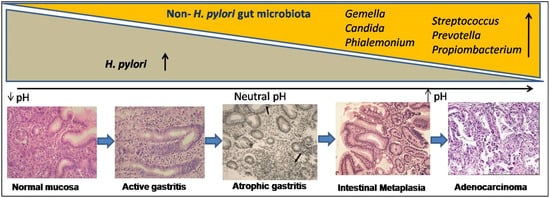
Metagenomics and advanced nucleotide sequencing techniques unhooked the mucosal and luminal composition of the gut microbiota and revealed their significance in natural habitats other than H. pylori [27][28][29][30]. In transgenic mice, gastrointestinal over-expression of human IL-1β was sufficient for the development of gastric dysplasia and carcinoma in a stepwise manner [31][32]. Some experimental animal models, particularly transgenic insulin-gastrin (INS-GAS) mice model, showed the importance of non- H. pylori gastric microbiome in enhancing the effects of H. pylori in the development of GC [33][34]. Patients with GC also showed dysbiotic microbial population with genotoxic potential that differs from the patients with chronic gastritis [35]. The H. pylori infection generates an inflammatory reaction in the stomach, resulting in the loss of parietal cells and an elevation in gastric pH. H. pylori may contribute to microbial dysbiosis, and effective eradication can restore the gut microbiota to a state comparable to that of uninfected people [36]. The colonization of gut microbiota grows over time as AG develops, and in spite of H. pylori becoming diminished, the precancerous lesionscontinue to develop ( Figure 2 ). Higher metabolites produced by dysbiosis of the gut microbiome, such as N-nitroso compounds and lactate, are thought to affect the immunological response as well as DNA damage, resulting in gastric carcinogenesis [37][38]. Recent studies proved that gut microbiota plays a significant role in the progression of gastric inflammation, AG, and IM after H. pylori eradication [39][40]. However, retrospective and only association-based findings are the limitations of these studies. It is unclear that the microbial changes seen in GC cause disease or are a result of the histologic progression through the precancerous cascade. Therefore, better understanding of the role of the gut microbiota in the development and progression of GC should lead to better diagnostic and preventive options.
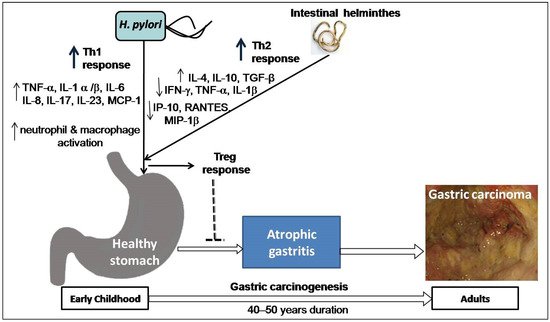
Co-infection with intestinal helminths may affect the outcome of H. pylori infection. A study on two Colombian communities infected with virulence-associated genotypes of H. pylori found a high versus low incidence of GC (endemic area of helminth infection), supporting the notion that intestinal helminth infection reduces the influence of H. pylori on gastric carcinogenesis [41]. Concurrent helminth infection has been shown in animal experiments to reduce the severity of H. pylori -induced gastritis [42]. Recently, a study on gastric mucosal samples also showed decreased expression of proinflammatory cytokines and predominant Th2 response (higher level IL-4) among H. pylori -infected humans co-infected with intestinal helminthes [43]. The higher load of H. pylori and intestinal parasites in the India and Venezuela populations is associated with a low risk of GC, which might explain these enigmas [9][43][44]. One study in a Chinese population found that concurrent helminth infections altered serological IgG responses as well as the pepsinogen I/II ratio, indicating a lower chance of developing H. pylori -induced atrophy [45]. A study on children from regions of low versus high risk of GC but similar H. pylori seropositivity showed that subjects from the low-risk area were more commonly infected with helminths and showed higher Th2-associated IgG1 responses to H. pylori infection [46]. These findings suggest that early childhood exposure to intestinal helminths induces immunoregulatory lymphocytes and anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β and lowers the expression of pro-inflammatory IFN-γ, TNF-α, and IL-1β, including Th1-associated IP-10, RANTES, and MIP-1β. Cysteinyl leukotrienes are produced by tuft cells during helminth infection [47]. On the other hand, chemosensing by tuft cells activates group 2 innate lymphoid cells (ILC2), leading to an increase in tuft cell frequency, and exhibiting significant physiologic alterations in the tissue, including hyperplasia of mucus-secreting goblet cells [48]. Intestinal remodeling and helminth removal require this feedback control pathway. This mechanism provides a new direction for future research towards co-infection of helminths and H. pylori . Another study showed that H. pylori infection causes diseases by hypermethylation of key cellular promoters at CpG dinucleotides (promoter silencing), especially in gastric mucosa [49]. However, helminths may induce pathological changes by epigenetic reprogramming of host cells [50]. Poor hygienic and environmental conditions favor the endemic prerequisite for transmission and existence of both H. pylori and intestinal helminths acquired at an early age. In particular, helminths promote a Th2-polarizing response that may decrease H. pylori -induced cancer risk in individuals later in life ( Figure 3 ). Studies addressing this issue are warranted and represent an intriguing area of research with the potential for future studies focused on H. pylori pathogenesis.
4. Inflammation-Related Genetic Polymorphisms Together with Other Factors as Initiators of Precancerous Lesions and GC
Chronic inflammation can occur in genetically susceptible hosts with defective mucosal host defense systems or dysregulated immune responses, leading to excessively aggressive responses to ubiquitous antigens, which is the root cause of inflammation-related carcinogenesis [20][23][51]. Inflammation-related genetic polymorphisms act as initiators of H. pylori -induced chronic atrophic gastritis, and together with other factors they become the precursor of precancerous lesions and carcinoma [52][53]. Cytokines induced by specific stimuli, such as toxins produced by pathogens, are involved in immunity, inflammation, and cell proliferation. By secreting cytokines and recruiting specific inflammatory cells, H. pylori influence both the mucosal and systemic immune responses. In addition, H. pylori cause cellular changes as well as changes in genes that are important for epigenetic integrity and mucosal homeostasis. These genetic changes during the development of chronic inflammation are the subject of extensive investigation. A primary strategy for screening and early detection of GC in individuals at risk may include finding persons who have H. pylori infection with a pro-inflammatory makeup [54]. Genetic variations in pro-inflammatory and anti-inflammatory cytokine genes influence individual response to carcinogenic exposures. The degree of inflammation in the host tissue is determined not only by external factors such as infection with bacteria but also on the host’s genetic makeup, whether they have a high- or a low-producing genotype.
Pro- and anti-inflammatory cytokines modulate the inflammatory response of the stomach mucosa [55][56]. Pro-inflammatory cytokines activate inflammatory cells by the migration of neutrophils, mononuclear phagocytes, eosinophils, and mast cells (e.g., IL-8, MCP-1, RANTES, TNF-α,and IL-1), and also play a significant role in acquired immune responses that can regulate the growth, differentiation, and activation of lymphocytes, mast cells, eosinophils, and other hemopoietic cells (e.g., IFN- γ, IL-12 etc.) [57]. In another way, anti-inflammatory cytokine (e.g., IL-10, a product of Th2 cells) is a potent factor for suppressing the inflammatory and neoplastic environment. It inhibits IFN- γ production and antigen-specific T-cell activation by down-regulating antigen presentation as well as IL-1, TNF- α, and IL-6 production by monocytes or macrophages [58]. Cytokines, particularly SNPs of IL-1B, IL-4, IL-6, IL-8, IL-10, IL17A, and IL-17F, may influence cancer prognosis and prevention [59]. A balance between pro- (IL-8) and anti-inflammatory (IL-10) cytokines may influence the degree of chronic inflammation, which is a potential factor in development of gastritis and GC [16][60]. Furthermore, genetic diversity of cytokine genes revealed differences in the severity of H. pylori -induced inflammation and the risk of GC among various populations. More data will be necessary to assess the above hypothesis.
Acid production, oxidative stress, and DNA damage are all affected by polymorphisms in both bacterial and host genes, which are thought to be a major factor in the pathogenesis of GC [61]. When H. pylori infect the gastric mucosa, it stimulates neutrophils and mononuclear cells, causing them to release a variety of inflammatory cytokines [62]. In particular, IL-1β and TNF-α are potent inhibitors of gastric acid secretion [55][63][64].They promote the development of non-H. pylori gut microbiota capable of sustaining inflammation and continually producing oxidative stress, thereby hastening the process of gastric carcinogenesis. Moreover, long-term H. pylori infection in the context of vulnerable host gene polymorphisms may result in hypochlorhydria or hyperchlorhydria, depending on the degree and location of gastritis, which may proceed to gastric ulceration or GC. However, we believe the integration of all the information will be helpful for future study into novel molecular pathways and processes implicated in H. pylori-induced inflammation.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Berardi, R.; Scartozzi, M.; Romagnoli, E.; Antognoli, S.; Cascinu, S. Gastric cancer treatment: A systematic review. Oncol. Rep. 2004, 11, 911–916.
- Plummer, M.; Franceschi, S.; Vignat, J. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490.
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori Infection and the Development of Gastric Cancer. N. Engl. J. Med. 2001, 345, 784–789.
- Hooi, J.K.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429.
- Kidd, M.; Lastovica, A.J.; Atherton, J.C. Heterogeneity in the Helicobacter pylori vacA and cagA genes: Association with gastroduodenal disease in South Africa? Gut 1999, 45, 499–502.
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740.
- Sugano, K. Premalignant conditions of gastric cancer. J. Gastroenterol. Hepatol. 2013, 28, 906–911.
- Ghoshal, U.C.; Chaturvedi, R.; Correa, P. The enigma of Helicobacter pylori infection and gastric cancer. Indian J. Gastroenterol. 2010, 29, 95–100.
- Correa, P.; Piazuelo, B.M.; Wilson, K. Pathology of Gastric Intestinal Metaplasia: Clinical Implications. Am. J. Gastroenterol. 2010, 105, 493–498.
- Wroblewski, L.E.; Peek, R.M.; Wilson, K. Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739.
- Singh, K.; Ghoshal, U.C. Causal role of Helicobacter pylori infection in gastric cancer: An Asian enigma. World J. Gastroenterol. 2006, 12, 1346–1351.
- Arnold, M.; Park, J.Y.; Camargo, M.C.; Lunet, N.; Forman, D.; Soerjomataram, I. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut 2020, 69, 823–829.
- Heer, E.V.; Harper, A.S.; Sung, H.; Jemal, A.; Fidler-Benaoudia, M.M. Emerging cancer incidence trends in Canada: The growing burden of young adult cancers. Cancer 2020, 126, 4553–4562.
- Terao, S.; Suzuki, S.; Yaita, H.; Kurahara, K.; Shunto, J.; Furuta, T.; Maruyama, Y.; Ito, M.; Kamada, T.; Aoki, R.; et al. Multicenter study of autoimmune gastritis in Japan: Clinical and endoscopic characteristics. Dig. Endosc. 2020, 32, 364–372.
- Kumar, S.; Kumari, N.; Mittal, R.D.; Mohindra, S.; Ghoshal, U.C. Association between pro-(IL-8) and anti-inflammatory (IL-10) cytokine variants and their serum levels and H. pylori-related gastric carcinogenesis in northern India. Meta Gene 2015, 6, 9–16.
- Shukla, S.K.; Prasad, K.N.; Tripathi, A.; Singh, A.; Saxena, A.; Ghoshal, U.C.; Krishnani, N.; Husain, N. Epstein-Barr virus DNA load and its association with Helicobacter pylori infection in gastroduodenal diseases. Braz. J. Infect. Dis. 2012, 15, 583–590.
- Valenzuela, M.A.; Canales, J.; Corvalan, A.; Quest, A.F.G. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 2015, 21, 12742–12756.
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545.
- Correa, P. Gastric cancer: Overview. Gastroenterol. Clin. N. Am. 2013, 42, 211–217.
- Mishra, K.K.; Srivastava, S.; Aayyagari, A.; Ghosh, K. Development of an animal model of Helicobacter pylori (Indian strain) infection. Indian J. Gastroenterol. 2019, 38, 167–172.
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867.
- Correa, P.; Schneider, B.G. Etiology of gastric cancer: What is new? Cancer Epidemiol. Biomark. Prev. 2005, 14, 1865–1868.
- Correa, P.; Haenszel, W.; Cuello, C.; Zavala, D.; Fontham, E.; Zarama, G.; Tannenbaum, S.; Collazos, T.; Ruiz, B. Gastric precancerous process in a high risk population: Cohort follow-up. Cancer Res. 1990, 50, 4737–4740.
- Correa, P.; Piazuelo, M. Natural history of Helicobacter pylori infection. Dig. Liver Dis. 2008, 40, 490–496.
- Pulipati, P.; Sarkar, P.; Jakkampudi, A. The Indian gut microbiota-Is it unique? Indian. J. Gastroenterol. 2020, 39, 133–140.
- Ghoshal, U.C.; Goel, A.; Quigley, E.M.M. Gut microbiota abnormalities, small intestinal bacterial overgrowth, and non-alcoholic fatty liver disease: An emerging paradigm. Indian J. Gastroenterol. 2020, 39, 9–21.
- Schulz, C.; Schütte, K.; Malfertheiner, P. Helicobacter pylori and Other Gastric Microbiota in Gastroduodenal Pathologies. Dig. Dis. 2016, 34, 210–216.
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The role of the gastric bacterial microbiome in gastric cancer: Helicobacter pylori and beyond. Ther. Adv. Gastroenterol. 2019, 12, 1756284819894062.
- Tu, S.; Bhagat, G.; Cui, G. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008, 14, 408–419.
- Huang, F.Y.; Chan, A.O.; Rashid, A. Interleukin-1beta increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol. Lett. 2016, 11, 2919–2924.
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of Commensal Flora in Helicobacter pylori–Infected INS-GAS Mice Reduces Gastritis and Delays Intraepithelial Neoplasia. Gastroenterology 2011, 140, 210–220.e4.
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47.
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2017, 67, 226–236.
- Guo, Y.; Zhang, Y.; Gerhard, M.; Gao, J.-J.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.-L.; Bajbouj, M.; Suchanek, S.; et al. Effect of Helicobacter pylori on gastrointestinal microbiota: A population-based study in Linqu, a high-risk area of gastric cancer. Gut 2019, 69, 1598–1607.
- Yang, J.; Zhou, X.; Liu, X.; Ling, Z.; Ji, F. Role of the Gastric Microbiome in Gastric Cancer: From Carcinogenesis to Treatment. Front. Microbiol. 2021, 12, 641322.
- Liu, X.; Shao, L.; Liu, X. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. E Bio. Med. 2019, 40, 336–348.
- Sung, J.J.Y.; Coker, O.O.; Chu, E.; Szeto, C.H.; Luk, S.T.Y.; Lau, H.; Yu, J. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after Helicobacter pylori eradication. Gut 2020, 69, 1572–1581.
- Barra, W.F.; Sarquis, D.P.; Khayat, A.S. Gastric Cancer Microbiome. Pathobiology 2021, 88, 156–169.
- Bravo, L.E.; van Doom, L.J.; Realpe, J.L. Virulence-associated genotypes of Helicobacter pylori: Do they explain the African enigma? Am. J. Gastroenterol. 2002, 97, 2839–2842.
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542.
- Fuenmayor-Boscán, A.; Hernández-Rincón, I.; Arismendi-Morillo, G.; Mengual, E.; Rivero, Z.; Romero, G.; Lizarzábal, M.; Álvarez-Mon, M. Changes in the severity of gastric mucosal inflammation associated with Helicobacter pylori in humans coinfected with intestinal helminths. Indian J. Gastroenterol. 2020, 39, 186–195.
- Hussain, Z.; El-Omar, E.; Lee, Y.Y. Dual infective burden of Helicobacter pylori and intestinal parasites: Good or bad news for the host? Indian J. Gastroenterol. 2020, 39, 111–116.
- Du, Y.; Agnew, A.; Ye, X.-P.; Robinson, P.A.; Forman, D.; Crabtree, J.E. Helicobacter pylori and Schistosoma japonicum co-infection in a Chinese population: Helminth infection alters humoral responses to H. pylori and serum pepsinogen I/II ratio. Microbes Infect. 2006, 8, 52–60.
- Whary, M.T.; Sundina, N.; Bravo, L.E.; Correa, P.; Quiñones, F.; Caro, F.; Fox, J.G. Intestinal Helminthiasis in Colombian Children Promotes a Th2 Response to Helicobacter pylori: Possible Implications for Gastric Carcinogenesis. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1464–1469.
- McGinty, J.W.; Ting, H.-A.; Billipp, T.E.; Nadjsombati, M.S.; Khan, D.M.; Barrett, N.A.; Liang, H.-E.; Matsumoto, I.; von Moltke, J. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-helminth Immunity in the Small Intestine but Are Dispensable for Anti-protist Immunity. Immunity 2020, 52, 528–541.e7.
- Von Moltke, J.; Ji, M.; Liang, H.E. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225.
- Yousefi, B.; Mohammadlou, M.; Abdollahi, M.; Farrokhi, A.S.; Karbalaei, M.; Keikha, M.; Kokhaei, P.; Valizadeh, S.; Rezaiemanesh, A.; Arabkari, V.; et al. Epigenetic changes in gastric cancer induction by Helicobacter pylori. J. Cell. Physiol. 2019, 234, 21770–21784.
- Minarovits, J. Microbe-induced epigenetic alterations in host cells: The coming era of patho-epigenetics of microbial infections. Acta Microbiol. Immunol. Hung. 2009, 56, 1–19.
- Okada, F.; Izutsu, R.; Goto, K.; Osaki, M. Inflammation-Related Carcinogenesis: Lessons from Animal Models to Clinical Aspects. Cancers 2021, 13, 921.
- Machado, J.C.; Figueiredo, C.; Canedo, P.; Pharoah, P.; Carvalho, R.; Nabais, S.; Alves, C.C.; Campos, M.L.; Van Doorn, L.-J.; Caldas, C.; et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology 2003, 125, 364–371.
- Graham, D.Y. Helicobacter pylori Update: Gastric Cancer, Reliable Therapy, and Possible Benefits. Gastroenterology 2015, 148, 719–731.e3.
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305.
- Sugimoto, M.; Furuta, T.; Yamaoka, Y. Influence of inflammatory cytokine polymorphisms on eradication rates ofHelicobacter pylori. J. Gastroenterol. Hepatol. 2009, 24, 1725–1732.
- White, J.R.; Winter, J.A.; Robinson, K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J. Inflamm. Res. 2015, 8, 137–147.
- Morse, H.; Olomolaiye, O.; Wood, N.; Keen, L.; Bidwell, J. Induced Heteroduplex Genotyping of Tnf-A, Il-1β, Il-6 And Il-10 Polymorphisms Associated With Transcriptional Regulation. Cytokine 1999, 11, 789–795.
- Bidwell, J.; Keen, L.; Gallagher, G.; Kimberly, R.; Huizinga, T.; McDermott, M.; Oksenberg, J.; McNicholl, J.; Pociot, F.; Hardt, C.; et al. Cytokine gene polymorphism in human disease: Online databases. Genes Immun. 1999, 1, 3–19.
- Yuzhalin, A. The role of interleukin DNA polymorphisms in gastric cancer. Hum. Immunol. 2011, 72, 1128–1136.
- Sugimoto, M.; Yamaoka, Y.; Furuta, T. Influence of interleukin polymorphisms on development of gastric cancer and peptic ulcer. World J. Gastroenterol. 2010, 16, 1188–1200.
- Smoot, D.T.; Elliott, T.B.; Verspaget, H.W.; Jones, D.; Allen, C.R.; Vernon, K.G.; Bremner, T.; Kidd, L.C.R.; Kim, K.S.; Groupman, J.D.; et al. Influence of Helicobacter pylori on reactive oxygen-induced gastric epithelial cell injury. Carcinogenesis 2000, 21, 2091–2095.
- Hwang, I.R.; Kodama, T.; Kikuchi, S. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology 2002, 123, 1793–1803.
- Wolfe, M.; Nompleggi, D. Cytokine inhibition of gastric acid secretion—A little goes a long way. Gastroenterology 1992, 102, 2177–2178.
- El-Omar, E.; Carrington, M.; Chow, W.-H.; McColl, K.E.L.; Bream, J.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.-C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402.


