Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Flora Guerra | + 4297 word(s) | 4297 | 2021-08-19 05:46:57 | | | |
| 2 | Vivi Li | Meta information modification | 4297 | 2021-08-31 08:12:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Guerra, F. RAB7 Protein. Encyclopedia. Available online: https://encyclopedia.pub/entry/13716 (accessed on 16 January 2026).
Guerra F. RAB7 Protein. Encyclopedia. Available at: https://encyclopedia.pub/entry/13716. Accessed January 16, 2026.
Guerra, Flora. "RAB7 Protein" Encyclopedia, https://encyclopedia.pub/entry/13716 (accessed January 16, 2026).
Guerra, F. (2021, August 30). RAB7 Protein. In Encyclopedia. https://encyclopedia.pub/entry/13716
Guerra, Flora. "RAB7 Protein." Encyclopedia. Web. 30 August, 2021.
Copy Citation
RAB7 is a small guanosine triphosphatase (GTPase) extensively studied as regulator of vesicular trafficking. Indeed, its role is fundamental in several steps of the late endocytic pathway, including endosome maturation, transport from early endosomes to late endosomes and lysosomes, clustering and fusion of late endosomes and lysosomes in the perinuclear region and lysosomal biogenesis. Besides endocytosis, RAB7 is important for a number of other cellular processes among which, autophagy, apoptosis, signaling, and cell migration. Given the importance of RAB7 in these cellular processes, the interest to study the role of RAB7 in cancer progression is widely grown.
RAB7
endocytosis
tumor progression
cisplatin resistance
extracellular vesicles
oncogene
oncosuppressor
1. Introduction
The RAB (Ras-related in brain) protein family comprises small guanosine triphosphatases (GTPases) that are important regulators of membrane identity and of vesicular trafficking events [1][2][3][4]. RAB7A is a small GTPase of the RAB family, ubiquitously expressed, mainly localized to late endosomes and with a pivotal role in endocytic trafficking. Indeed, RAB7A regulates maturation of early endosome into late endosome, transport from early endosomes to late endosome and lysosomes, clustering and fusion of late endosomes and lysosomes in perinuclear region and lysosomal biogenesis [5][6]. In addition of these functions, and related to them, RAB7A has many other cellular roles, being involved in autophagy [7][8], in apoptosis [9], in phagocytosis [10], in retromer regulation, mitophagy, and lipophagy as well as in cytoskeleton organization [5]. RAB7A plays specific key roles in neurons as a regulator of neurotrophin receptor trafficking and signaling, neuritogenesis, axonal retrograde transport, and neuronal migration [5][11][12][13]. Furthermore, RAB7A has been involved in host-pathogen interaction as often microbial or viral pathogens target this protein or its regulators or effector to subvert phagocytosis or other cellular processes [14][15] but also in thyroid hormone production [16] and extracellular vesicle secretion [17].
As all GTPases, RAB7A functions are guaranteed by a cyclical mechanism of activation and inactivation, that depends on GTP binding and hydrolysis, thus shuttling between active GTP-bound and inactive guanosine diphosphate (GDP)-bound state. A number of proteins control this cycle and, among them, the most relevant are the Guanine nucleotide Exchange Factors (GEFs) that stimulate GDP dissociation and GTP acquisition and GTPase-Activating Proteins (GAPs) that are required to prompt GTP hydrolysis. In addition, the Rab Escort Proteins (REPs) recognize newly synthesized RAB7A proteins and present them to the prenylation enzyme Rab geranylgeranyl transferase (RabGGT) allowing geranyl-geranylation, useful for anchorage to membranes. In the cytoplasm, GDP-bound geranyl-geranylated RAB7A is associated to GDP Dissociation Inhibitor (GDI), while GDI displacement is operated by the GDI Displacement Factor (GDF). After GDI displacement, RAB7A is recruited to membranes where it is activated by GEFs through nucleotide exchange [18][19]. Active (GTP-bound) RAB7A is able to interact with several different effector proteins, through which it regulates a number of downstream functions [20]. Two mutants of RAB7 are widely used to study the function of this GTPase: a dominant negative mutant (RAB7T22N) characterized by impaired nucleotide exchange, thus locking the protein in its inactive GDP-bound form, and a constitutively active mutant (RAB7Q67L) characterized by impaired GTP hydrolysis, thus locking the protein in its active GTP-bound form [5].
Considering the involvement of RAB7A in numerous cellular processes, it is easy to imagine that alterations of its expression, biochemical properties, such as GTP exchange and hydrolysis, interaction with effectors and mutations may cause occurrence of pathological conditions. Indeed, for instance, mutations in the RAB7A gene cause the Charcot-Marie-Tooth type 2B (CMT2B) peripheral neuropathy [21][22][23][24][25][26] and there are numerous reports in the literature indicating RAB7A as a lead actor of cancer [5].
2. RAB7A in Cancer Progression
The tumor pathogenesis is a multistep process characterized by progressive evolvement of normal cells to the neoplastic state acquiring the traits to become tumorigenic and, ultimately, malignant [27][28].
During cancer progression the cells acquire in succession the following capabilities: (i) Sustaining proliferative signaling, (ii) evading growth suppressors, (iii) resisting cell death, (iv) enabling replicative immortality, (v) inducing angiogenesis, (vi) activating invasion and metastasis, (vii) reprogramming of energy metabolism, and (viii) evading immune destruction [29]. Genome instability and inflammation are at the base of these hallmarks. Indeed, acquisition of these hallmarks is expedited by generation of genetic diversity and is promoted by inflammation. Moreover, a further aspect that should not be ignored is that normal cells contribute to the acquisition of the eight hallmarks creating the “tumor microenvironment” [29]. For example, epithelial–mesenchymal transition (EMT) is the process by which cancer cells change their phenotype from epithelial to mesenchymal to become invasive and to start metastatization. This aspect of cancer progression is supported by cancer-associated fibroblasts (CAFs) in the stroma that surrounds the growing tumor mass [29].
In the context of cancer progression, genes that determine gain of functions inducing abnormal cellular growth to favor tumorigenesis are known as oncogenes, while tumor suppressors genes protect cells from cancer progression until loss of heterozygosis (LOH) [30]. According to these definitions, RAB7A was described as an oncogene or tumor suppressor depending on its capacity to favor tumor progression or to prevent malignant growth, inducing or blocking different aspects within the eight hallmarks of cancer. The different impact of RAB7A in cancer progression, probably depending on the cellular context and on other environmental factors, is analyzed below.
2.1. Oncogenic Functions of RAB7A
EMT is characterized by several molecular changes, including downregulation of epithelial markers and key components of intercellular junctions, such as E-cadherin, claudins, and occludins, disruption of desmosomes as well as upregulation of the mesenchymal markers N-cadherin, vimentin and fibronectin, thus fostering motility and invasion. EMT allows the cells to gain migratory and invasive features through degradation of extracellular matrix (ECM) proteins, actin cytoskeleton reorganization, formation of membrane protrusions required for invasive growth, extravasation, angiogenesis, as well as anoikis evasion and drug resistance [31][32].
Recently, it was discovered that RAB7A is important for actin cytoskeleton organization [33][34][35]. Indeed, RAB7 interacts with RAC1, a small GTPase involved in the regulation of actin cytoskeleton [36]. Overexpression of RAB7 increases RAC1 activity, while RAB7 silencing caused RAC1 inactivation [36]. Moreover, ARMUS, an effector of RAC1, is a TBC RAB-GAP that inactivates RAB7 coordinating RAB7 and RAC1 functions during autophagy [34][35].
Among several RAB7A effectors, there are two members of the intermediate filament (IFs) proteins, vimentin, and peripherin. IFs, one of the three components of the cytoskeleton, are constituted through the assembly of soluble precursors into insoluble protein polymers [37][38]. IFs not only are the major determinants of cell architecture, but also regulation of membrane trafficking and organelle positioning and function dependent on them [39]. Furthermore, it is known that upregulation of vimentin in epithelial cells favors acquisition of the mesenchymal phenotype increasing cell motility, inducing physical changes in cell shape, loss of cell–cell contacts, and increasing the turnover of focal adhesions [40][41]. Coherently, loss of vimentin in mesenchymal MDA-MB-231 cells significantly decreases the ability of these cells to migrate and invade [42]. Furthermore, RAB7A is able to regulate cell migration through Ras-related C3 botulinum toxin substrate 1 (RAC1) and vimentin in human lung cancer NCI H1299 cells [43]. Direct interaction between vimentin and RAB7A allows RAB7A-mediated regulation of vimentin filament assembly [44]. Indeed, vimentin phosphorylation is increased by overexpression of RAB7Awt or of the constitutively active RAB7AQ67L mutant, with consequent redistribution of vimentin in the soluble fraction, while decreased of vimentin phosphorylation and increased amount of filamentous vimentin was determined by RAB7A depletion [44]. Moreover, reorientation of vimentin during migration is determined by RAB7A [43]. RAB7A depletion strongly affects cell velocity and directness during migration, determining alteration of β1-integrin activation, distribution, and trafficking and thus hampering cell adhesion and spreading [43]. Furthermore, RAB7A depletion reduces RAC1 activation, interfering with filopodia formation and, altering vimentin organization [43].
Tumor-necrosis factor-α (TNF-α) and TNF-β are among stimuli required to induce EMT [45]. After stimulation with TNF-α to induce EMT and invasiveness, RAB7A expression was increased in cholangiocarcinoma cells [45]. Also in thyroid adenomas and in ovarian/primary peritoneal serous carcinoma overexpression of RAB7A was observed in cancerous tissues [16][46].
In highly aggressive tumors, such as ovarian, melanoma, breast and colorectal the capacity of primary tumor cells to invade the surrounding tissues through ECM degradation is linked to the expression of matrix metalloproteinases (MMPs) [47][48][49][50][51]. Furthermore, membrane-anchored membrane type 1 matrix metalloproteinase (MT1-MMP) has a central role in this process [49][52][53][54]. To this purpose, MT1-MMP induces degradation of ECM through its proteolytic function determining cleavage of collagen types I, II, and III, laminins 1 and 5, fibronectin, vitronectin, fibrin, and aggrecan, but also it is able to activate pro-MMPs [55]. Once activated, MT1-MMP is transported to the plasma membrane in order to mediate ECM proteolytic degradation and to favor cell invasion. Proteolytic activity of MT1-MMP is reduced by internalization [56][57], but internalization causes also recycling of MT1-MMP back to the plasma membrane and therefore it is essential for MT1-MMP degradative function [58]. It has been demonstrated in cervical carcinoma HeLa and in fibrosarcoma HT-1080 cells that endosomal trafficking and recycling of MT1-MMP1 are dependent on RAB7A and VAMP7 [59]. Interestingly, inhibition of recycling, using dominant negative mutants of RAB7 and VAMP7, reduces both invasion and migration [59]. Thus, it has been demonstrated a role of RAB7A in the recycling pathway, which is important for RAB7A oncogenic functions [59].
Epidermal Growth Factor Receptor (EGFR) and Human Epidermal growth factor Receptor 2 (HER2) are members of receptor tyrosine kinases (RTKs) ErbB family. High expression of both proteins is detected in breast cancer and it is associated with more aggressive cancer phenotypes and poorer prognosis [60]. Indeed, RTKs are activated by ligand binding with subsequent dimerization, internalization and initiation of pro-oncogenic intracellular signaling via mitogen-activated protein kinase (MAPK) and PI3K/Akt pathways. Thus, enhanced expression/activation of EGFR or HER2 determines aberrant expression of genes involved in cancer proliferation, survival, migration, and angiogenesis [61]. Endocytosis-mediated internalization of ligand-bound RTKs is an important process for the regulation of downstream signaling [62]. Indeed, activated receptors, after internalization, are sorted in early endosomes and subsequently recycled to the plasma membrane or degraded [63]. The recycling of receptors allows maintenance of activated receptor number on plasma membrane and is responsible for prolongated signaling [63]. RAB7 is essential for the degradation of signaling receptors, being responsible of sorting them into late endosome and of their transport to lysosomes [64]. Indeed, for instance, in HeLa cells the kinetic of EGFR degradation is accelerated or slowed by expressing the constitutively active or the dominant negative mutant of RAB7, respectively [65]. Intriguingly, it was demonstrated that, in human cervix squamous carcinoma A431 and breast cancer MCF7 cells, RAB7 is responsible for Akt survival signal maintenance during cell detachment or when HSP90 is inhibited, protecting cells from apoptosis [66]. In fact, RAB7 silencing suppresses anchorage-independent growth with a concomitant increase of anoikis and a reduction of active phosphorylated Akt [66]. In agreement, in breast cancer cells, the RAB7 interactor Rabring 7 was discovered to have a key role [67][68]. Accordingly, the gene was renamed Breast Cancer Associated gene 2 (BCA2) [67][68]. Overexpression of BCA2 in HeLa cells causes failure of RAB7 membrane association and thus RAB7 remains in the cytosol, not being able to carry out its functions on the endosomal membranes [69]. As a consequence, EGFR degradation is inhibited [69].
Phosphatase and tensin homolog (PTEN) is an important tumor suppressor, mutated in various types of cancers [70]. As a dual specific phosphatase, PTEN acts on both lipid and protein substrates being responsible for suppression of EGFR-mediated cell growth and proliferation signaling [71][72]. Moreover, phosphatidylinositol-3,4,5-trisphosphate (PIP3) is converted into phosphatidylinositol-4,5-bisphosphate (PIP2) at the cellular membrane by PTEN, which also negatively regulates oncogenic PI3K-AKT signaling and is associated with PI(3)P-containing endosomes [73]. RAB7 S72 and Y183 residues are essential for maturation of late endosomes. Indeed, both residues guarantee RAB7-GDI association, subsequent delivery to late endosomal membranes and activation by the Mon1a–Ccz1 GEF complex [74][75]. Recent work has discovered in HeLa and in human embryonic kidney HEK 293T cells a PTEN-dependent regulation of RAB7, strengthening the link between RAB7 and cancer [74]. PTEN controls EGFR signaling through activation of RAB7-mediated endosome maturation, acting on S72 and Y183 residues of RAB7 [74]. Mutations of PTEN are very frequent in several tumors [76][77]. Notably, the authors demonstrated that PTEN mutation at residue 138, determining oncosuppressor inactivation, affects RAB7 dephosphorylation at S72 and Y183 residues, with loss of the control of RAB7-dependent endosomal degradation and consequent uninterrupted growth signaling with important implications for tumor progression [74].
Cancer proliferation can arise also when immune surveillance is compromised and unchecked tumor cells proliferate and grow to form tumors [78]. Indeed, inflammatory response has a pivotal role in different stages of cancer progression, including initiation, promotion, malignant phenotype acquisition, invasion and metastasis [79][80]. Myeloid-derived suppressor cells (MDSCs) expansion is one of manifestations of inflammations [81]. MDSCs are able to suppress immune surveillance and directly induce tumor cell proliferation in vitro, and tumor growth and invasion in vivo [82][83][84][85]. Metabolic reprogramming and evading immune destruction are two of cancer important hallmarks. Interestingly, metabolic reprogramming of MDSCs is regulated by Lysosomal Acid Lipase (LAL). LAL generates free cholesterol and free fatty acids in the lysosomes through hydrolysis of cholesteryl esters and triglycerides [84]. LAL deficient (lal−/−) MDSCs compensate energetic deficit through increase glycolytic metabolism, ATP production, reactive oxygen species (ROS) over-production and overactivation of mammalian target of rapamycin (mTOR) [84]. In lal−/− MDSCs, development, systemic expansion, trans-endothelial migration, immune suppression and direct stimulation of tumor cell proliferation are regulated by mTOR [80][82][83][84][86][87]. RAB7 interacts with mTOR and is upregulated in lal−/− MDSCs [80][88]. A direct role of RAB7 in the regulation of MDSCs metabolism was demonstrated [80][88]. In fact, depletion of RAB7A in lal−/− MDSCs reduced overactivation of mTOR, decreased glucose consumption and ROS over-production, and increased the number of healthy mitochondria [80][88]. Moreover, it was demonstrated that RAB7 knockdown reduced lal−/− MDSCs differentiation from bone marrow, decreased trans-endothelial migration and abrogated T cell suppression [88]. Furthermore, RAB7 knockdown reduced the ability of MDSCs to induce tumor cell proliferation in vitro, tumor growth and tumor invasion in vivo [88]. Similar to what was observed in lal−/− MDSCs, RAB7 is upregulated also in lal−/− endothelial cells (ECs) [89]. Also, stimulation of RAB7 activity determines the ability of lal−/− ECs to stimulate tumor cell proliferation and metastasis [89]. Importantly, inhibition of RAB7 in lal−/− ECs was able to revert the tumor phenotype causing reduction of their enhanced migration and suppressing in vitro proliferation and in vivo tumor growth and metastasis [89].
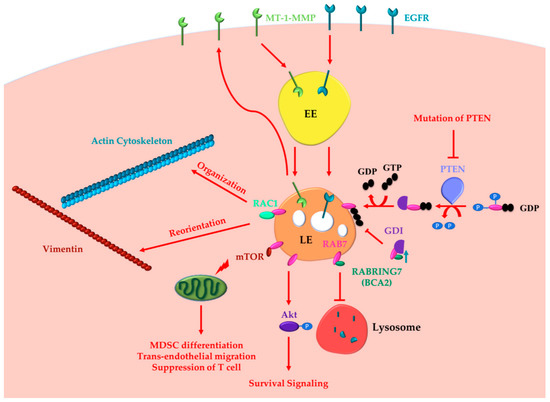
Altogether these data demonstrate the oncogenic functions of RAB7 (Figure 1).
Figure 1. RAB7 (Ras-related in brain 7) oncogenic functions in cancer progression. RAB7A determines the acquisition of invasion and metastasis features through different routes: (i) inducing actin cytoskeleton organization and vimentin reorientation thanks to RAC1 (Ras-related C3 botulinum toxin substrate 1) interaction; (ii) determining internalization and recycling of MT-1-MMP to allow extracellular matrix degradation. Other cancer hallmarks, such as metabolic reprogramming, evasion from immune destruction and angiogenesis, are induced by RAB7 through interaction with mTOR (mammalian target of rapamycin). Indeed, RAB7 induces activation of mammalian target of rapamycin (mTOR) decreasing the number of healthy mitochondria and inducing MDSC (myeloid-derived suppressor cells) differentiation, trans-endothelial migration, and T cell suppression. RAB7 induces Akt-mediated survival signaling. RAB7 interacts with Rabring7, also named Breast Cancer Associated gene 2 (BCA2), and inhibits Epidermal Growth Factor Receptor (EGFR) degradation, while BCA2-overexpression determines RAB7 sequestration in the cytosol by the GDP (guanosine diphosphate) Dissociation Inhibitor (GDI), and prevents EGFR degradation. Also, PTEN (phosphatase and tensin homologue) mutations, determining lack of RAB7 dephosphorylation and impairment of endosomal maturation, inhibit EGFR degradation. These latter oncogenic functions of RAB7 are involved in the acquisition of three cancer hallmarks: resisting cell death, evading growth suppressors and sustained proliferative signaling. EE, Early Endosome; LE, Late Endosome.
2.2. Oncosuppressor Functions of RAB7A
An important obstacle to neoplastic transformation is represented by growth factor dependence. Indeed, lack of growth factors induces withdrawal of cells from the cell cycle and activation of apoptosis. Growth factors induce cell proliferation but also suppression of intrinsic mitochondria-mediated apoptosis [90]. In fact, deprivation of growth factors induces rapidly a decline of the rate of glucose and amino acid uptake, and loss of the receptors responsible for the cellular uptake of iron (transferrin receptor) and cholesterol (LDL receptor) from the surface of the cells [91][92][93][94]. These metabolic alterations induce destabilization of mitochondrial physiology and bioenergetic dysfunction with consequent decrease of mitochondrial membrane potential and final release of proapoptotic mediators [95][96].
Tumor cells may acquire advantageous mutations to gain growth factor independence, to avoid apoptosis and to proliferate in the absence of extrinsic signals. Growth factor independent survival is supported by the activated form of the oncogene AKT, which maintains high rate of glycolysis, preserves mitochondrial membrane potential and prevents apoptosis [91]. It was demonstrated that RAB7 has a role in growth factor independent survival [91]. In fact, degradation of nutrient transporters is prevented by inhibition of RAB7 despite growth factor withdrawal [91]. Indeed, in murine prolymphocytic FL5.12 RAB7-depleted cells, proteins that have entered the endocytic pathway and that normally would be degraded in lysosomes, are instead recycled to the plasma membrane and re-expressed on the cell surface [91]. Thus, during growth factor withdrawal expression of the dominant negative mutant RAB7T22N causes maintenance of the mitochondrial membrane potential [90]. Also, in TP53 (Tumor Protein p53) null primary mouse embryonic fibroblasts, expression of the RAB7T22N dominant negative mutant cooperates with the adenoviral protein E1A, which inhibits Rb (retinoblastoma tumor suppressor), in order to promote cancer transformation [90]. Accordingly, it was established that RAB7 activity is regulated by growth factor availability in murine prolymphocytic FL5.12 cells [97]. In fact, during nutrient deprivation, RAB7 moves from the cytosol to the late endosomal and lysosomal membranes with a consequent consistent increase of the GTP-bound form [97]. Moreover, expression of the constitutively active RAB7Q67L mutant, which displays impaired GTPase activity and is mostly GTP-bound, is sufficient to trigger apoptosis even in the presence of growth factors and it is also able to reverse independent growth factor survival induced by protein kinase C (PKC) δ inhibition [97].
Oncosuppressor-like functions of RAB7 have also been described in prostate cancer [98]. It is known that in human prostate cancer DU-145 cells the intracellular localization of lysosomes generally closer to the cell surface determines the secretion of proteases favoring cell invasion [98][99]. Instead, perinuclear localization of lysosomes is a common feature of less invasive cells, which do not usually secrete large amounts of acid hydrolases. The movement of lysosomes in the cells are guaranteed by microtubules and actin filaments utilizing molecular motor protein such as dynein, kinesin, and/or myosin family members [100]. In addition, several GTPases, among which RAB7, regulate motor protein activity determining the intracellular distribution of lysosomes and other organelles in the cell [5]. The reported RAB7 role as a tumor suppressor is based on the fact that it is a negative regulator of prostate cancer growth and invasion because it inhibits ligand-induced c-Met signaling, known to induce EMT, and it controls the perinuclear localization of lysosomes [98]. Indeed, depletion of RAB7 strongly increases secretion of proteases, supporting larger tumors formation in vivo and increased invasive capability into surrounding tissue [98]. Troglitazone is an agonist of the peroxisome proliferator-activated receptor-γ (PPAR-γ), used for the treatment of type II diabetes because of its ability to improve insulin sensitivity [101]. Interestingly, this compound has several PPAR-γ-independent effects and, for instance, influences cell migration and invasion in several malignancies [102]. Indeed, Troglitazone and other members of the Thiazolinedione family inhibit RAB7-induced lysosome trafficking towards the cell surface and consequent Cathepsin B secretion [98].
The RAB7 behavior as a tumor suppressor has been demonstrated also in glioblastoma [103]. Extensive activation of RTKs was found in greater than 45% of gliobastoma specimens harboring EGFR amplification or mutations [104]. CD44 is a cell surface receptor responsible for increasing RTKs signaling and is defined also as co-receptor. CD44 is characterized by extensive alternative splicing that produces two main families of isoforms, termed CD44v and CD44s [105]. The role of the CD44s splice isoform has been studied and it was discovered that it is involved in attenuation of endocytosis-mediated EGFR degradation and in sustaining downstream Akt signaling [103]. Interestingly, CD44s interacts with RAB7A and blocks RAB7-mediated EGFR degradation by stimulating GTP hydrolysis in glioblastoma multiform cell lines [103].
Several RAB7-GAP have been identified and among them there is the phospho-protein folliculin (FLCN) [106]. Mutations of the FLCN gene are the cause of Birt–Hogg–Dubé disease characterized by the development of renal cell cancer, benign skin lesions, and lung cystis [107]. RAB7 interacts with FLCN and together they are able to regulate EGFR signaling [106]. Indeed, in FLCN-deficient thyroid cancer FTC-133 cells, dysregulation of RAB7 activity due to loss of FLCN determined slower EGFR degradation, increased EGFR signaling, and tumor growth [106].
Finally, RAB7 is activated by Liver Kinase B1 (LKB1), a serine–threonine protein kinase of calcium calmodulin family, ubiquitously expressed in several tissues, including liver, heart, lung, and skeletal muscle [108]. Hypoxia determines LKB1 nuclear export to the cytosol where it interacts with the Neuropilin-1 (NRP-1) protein and with RAB7 [108]. NRP-1 is a receptor of the Vascular Endothelial Growth Factor (VEGF) and its enhanced expression during tumorigenesis determines the angiogenetic switch with a role in cell survival, migration and invasion [109][110][111]. LKB1 binds to the constitutively active RAB7Q67L mutant protein but not to the dominant negative RAB7T22N mutant protein, indicating that it binds preferentially to the GTP-bound form of RAB7 [108]. LKB1 has been identified in lung cancer A549 cell lines as a RAB7 effector that alters NRP-1 trafficking causing NRP-1 localization to lysosomes and subsequent NRP-1 degradation [108]. The lysosomal degradation of NRP-1 causes decreased tumor angiogenesis and decreased tumor growth in vivo [108]. In contrast, depletion of RAB7 by RNA interference blocks transfer of NRP-1 to lysosomes, causing increased NRP-1 expression, increased angiogenesis and increased tumor growth [108].
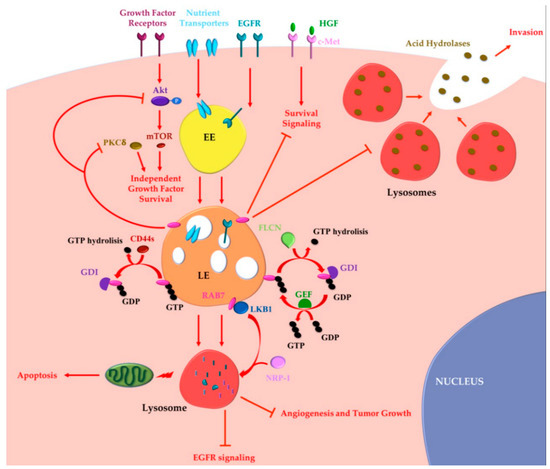
Altogether these data indicate that RAB7 can also exert oncosuppressor functions (Figure 2).
Figure 2. RAB7 oncosuppressor functions in cancer progression. Tumor cells may acquire growth factor independence in order to avoid apoptosis and to proliferate in the absence of extrinsic signals. RAB7 is able to reverse independent growth factor through inhibition of Akt and PKCδ-regulated survival signaling. Moreover, during growth factor withdrawal, nutrient transporters are removed from the cell surface and degraded by lysosomes in a RAB7-regulated process. Nutrient transporters degradation determines impairment of cellular bioenergetics, loss of mitochondrial homeostasis and, finally, activation of apoptosis preventing growth factor-independent tumor proliferation. RAB7 is essential for EGFR degradation and for removal of sustained proliferative signals. EGFR degradation is also guaranteed by GAP (GTPase-Activating Protein) FLCN (folliculin) activity, which allows cyclical RAB7 regulation. CD44s inhibits RAB7 by inducing GTP hydrolysis and attenuation of endocytosis-mediated EGFR degradation, underling the importance of RAB7 in EGFR degradation. Liver Kinase B1 (LKB1) is an effector of RAB7, which induces Neuropilin-1 (NRP-1) lysosomal localization and degradation with consequent decreased tumor angiogenesis and tumor growth. RAB7 is also a negative regulator of the Hepatocyte Growth Factor (HGF)-Met signaling axis and controls the perinuclear localization of lysosomes avoiding lysosomal peripheral localization, acid hydrolases secretion and cancer invasion. EE, Early Endosome; LE, Late Endosome; GEF, GTP Exchange Factor; GDI, GDP Dissociation Inhibitor.
2.3. Is RAB7A an Oncojanus?
Gasparre and coworkers have coined the name oncojanus to identify a novel-type of tumor-implicated genes that have the ability to behave either as oncogenes or oncosuppressor during tumorigenesis [112]. In fact, they discovered that for a number of mitochondrial genes of which both oncogenic and suppressor roles have been demonstrated, tumor growth arrest is induced when disruptive mutations reach a critical mutant load [112][113]. In contrast, nondisruptive or below threshold mutations in the same genes prompt tumor growth [112][113].
Interestingly, also the RAB7A gene can behave as an oncojanus with oncogenic or oncosuppressor roles. Indeed, expression levels of RAB7 change during melanoma tumor progression [114]. Initially, in benign nevi, RAB7 is expressed at low levels under regulation of the transcription factor Sex determining region Y (SRY)-related High Motility Group (HMG)-box 10 (SOX10) [114]. Then, in primary non-invasive melanomas, RAB7 expression is strongly increased and under the control of the MYC oncogene [114]. These observations have suggested role of RAB7 in benign nevi transformation into melanoma. However, in primary invasive melanomas, RAB7 expression is strongly reduced, although never abolished, compared to non invasive ones [114]. Thus, RAB7 downregulation at this stage seems to important for acquisition of invasive properties [114]. In addition, downregulation of RAB7 correlates with increased risk of development of metastasis [114]. Finally, in melanoma metastasis expression of RAB7 seems to increase compared to primary invasive melanomas [114]. Thus, RAB7 expression varies during tumor progression suggesting that in the first step of transformation from benign nevi to non-invasive melanoma RAB7 behaves as an oncogene while in the following progression to invasive melanoma it could as a suppressor and therefore is downregulated.
RAB7 seems to behave as an oncojanus also in inflammatory breast cancer. Inflammatory breast cancer is a lethal form of breast cancer characterized by excessive lymphovascular invasion, which is responsible for metastatic dissemination, resistance to chemotherapy and cancer recurrence [115]. MARY-X is a human xenograft model of inflammatory breast cancer representing a good tool to investigate this kind of cancer as it forms spheroids that have biological similarities with the lymphovascular embolus but also with the blastocyst [116]. In the lymphovascular embolus, active clonal selection in the main tumor mass takes place and the stem cell phenotype emerges, exhibiting enhanced stem cell signaling and survival pathways [117][118]. Only a fraction of MARY-X become lymphovascular emboli in vivo and only a fraction of MARY-X cells forms spheroids in vitro. Nevertheless, sequential events of MARY-X spheroidogenesis in vitro have been characterized and include modulation of RAB7 expression and RAB7-regulated production of different forms of E-cadherin [119]. Besides the fact that modulation of RAB7 expression by silencing or overexpression alters spheroidogenesis events, it is interesting to note that the first steps of spheroidogenesis, the early aggregate-to-spheroid transition stages, were characterized by downregulation of RAB7 expression while the well-formed compact spheroids were characterized by strong RAB7 expression [119]. Therefore, also in this case, RAB7 expression follows a complex pattern as initially down-regulation of RAB7 is required for the formation of the loose spheroids but then high RAB7 expression is required to form compact and well-formed spheroids [119], suggesting that RAB7 behaves as an oncojanus.
References
- Pfeffer, S.R. Rab gtpases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715.
- Pfeffer, S.R. Rab gtpase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419.
- Zhen, Y.; Stenmark, H. Cellular functions of rab gtpases at a glance. J. Cell Sci. 2015, 128, 3171–3176.
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525.
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34.
- Langemeyer, L.; Frohlich, F.; Ungermann, C. Rab gtpase function in endosome and lysosome biogenesis. Trends Cell Biol. 2018, 28, 957–970.
- Kuchitsu, Y.; Fukuda, M. Revisiting rab7 functions in mammalian autophagy: Rab7 knockout studies. Cells 2018, 7, 215.
- Wen, H.; Zhan, L.; Chen, S.; Long, L.; Xu, E. Rab7 may be a novel therapeutic target for neurologic diseases as a key regulator in autophagy. J. Neurosci. Res. 2017, 95, 1993–2004.
- Snider, M.D. A role for rab7 gtpase in growth factor-regulated cell nutrition and apoptosis. Mol. Cell 2003, 12, 796–797.
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for rab7. Biochim. Biophys. Acta 2013, 1833, 503–510.
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small gtpase rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor trka. J. Neurosci. 2005, 25, 10930–10940.
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305.
- Kawauchi, T.; Sekine, K.; Shikanai, M.; Chihama, K.; Tomita, K.; Kubo, K.; Nakajima, K.; Nabeshima, Y.; Hoshino, M. Rab gtpases-dependent endocytic pathways regulate neuronal migration and maturation through n-cadherin trafficking. Neuron 2010, 67, 588–602.
- Mottola, G. The complexity of rab5 to rab7 transition guarantees specificity of pathogen subversion mechanisms. Front. Cell Infect. Microbiol. 2014, 4, 180.
- Wozniak, A.L.; Long, A.; Jones-Jamtgaard, K.N.; Weinman, S.A. Hepatitis c virus promotes virion secretion through cleavage of the rab7 adaptor protein rilp. Proc. Natl. Acad. Sci. USA 2016, 113, 12484–12489.
- Croizet-Berger, K.; Daumerie, C.; Couvreur, M.; Courtoy, P.J.; van den Hove, M.F. The endocytic catalysts, rab5a and rab7, are tandem regulators of thyroid hormone production. Proc. Natl. Acad. Sci. USA 2002, 99, 8277–8282.
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of rab7a protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52.
- Cherfils, J.; Zeghouf, M. Regulation of small gtpases by gefs, gaps, and gdis. Physiol. Rev. 2013, 93, 269–309.
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877.
- Wang, T.; Ming, Z.; Xiaochun, W.; Hong, W. Rab7: Role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal. 2011, 23, 516–521.
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of rab7 mutant proteins associated with charcot-marie-tooth type 2b disease. J. Neurosci. 2008, 28, 1640–1648.
- De Luca, M.; Bucci, C. A new v-atpase regulatory mechanism mediated by the rab interacting lysosomal protein (rilp). Commun. Integr. Biol. 2014, 7, 1–4.
- Meggouh, F.; Bienfait, H.M.; Weterman, M.A.; de Visser, M.; Baas, F. Charcot-marie-tooth disease due to a de novo mutation of the rab7 gene. Neurology 2006, 67, 1476–1478.
- Houlden, H.; King, R.H.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel rab7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590.
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small gtp-ase late endosomal protein rab7 cause charcot-marie-tooth type 2b neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727.
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel rab7 mutation in a chinese family with charcot-marie-tooth type 2b disease. Gene 2014, 534, 431–434.
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799.
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction: A novel potential driver of epithelial-to-mesenchymal transition in cancer. Front. Oncol. 2017, 7, 295.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Mascia, A.; Gentile, F.; Izzo, A.; Mollo, N.; De Luca, M.; Bucci, C.; Nitsch, L.; Calì, G. Rab7 regulates cdh1 endocytosis, circular dorsal ruffles genesis and thyroglobulin internalization in a thyroid cell line. J. Cell Physiol. 2016, 231, 1695–1708.
- Carroll, B.; Mohd-Naim, N.; Maximiano, F.; Frasa, M.A.; McCormack, J.; Finelli, M.; Thoresen, S.B.; Perdios, L.; Daigaku, R.; Francis, R.E.; et al. The tbc/rabgap armus coordinates rac1 and rab7 functions during autophagy. Dev. Cell 2013, 25, 15–28.
- Frasa, M.A.; Maximiano, F.C.; Smolarczyk, K.; Francis, R.E.; Betson, M.E.; Lozano, E.; Goldenring, J.; Seabra, M.C.; Rak, A.; Ahmadian, M.R.; et al. Armus is a rac1 effector that inactivates rab7 and regulates e-cadherin degradation. Curr. Biol. 2010, 20, 198–208.
- Sun, Y.; Buki, K.G.; Ettala, O.; Vaaraniemi, J.P.; Vaananen, H.K. Possible role of direct rac1-rab7 interaction in ruffled border formation of osteoclasts. J. Biol. Chem. 2005, 280, 32356–32361.
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783.
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771.
- Styers, M.L.; Kowalczyk, A.P.; Faundez, V. Intermediate filaments and vesicular membrane traffic: The odd couple’s first dance? Traffic 2005, 6, 359–365.
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883.
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851.
- Messica, Y.; Laser-Azogui, A.; Volberg, T.; Elisha, Y.; Lysakovskaia, K.; Eils, R.; Gladilin, E.; Geiger, B.; Beck, R. The role of vimentin in regulating cell invasive migration in dense cultures of breast carcinoma cells. Nano Lett. 2017, 17, 6941–6948.
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through rac1 and vimentin. Biochim. Biophys. Acta 2017, 1864, 367–381.
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small gtpase rab7a. Biochim. Biophys. Acta 2013, 1833, 1283–1293.
- Suwandittakul, N.; Reamtong, O.; Molee, P.; Maneewatchararangsri, S.; Sutherat, M.; Chaisri, U.; Wongkham, S.; Adisakwattana, P. Disruption of endocytic trafficking protein rab7 impairs invasiveness of cholangiocarcinoma cells. Cancer Biomark. 2017, 20, 255–266.
- Davidson, B.; Zhang, Z.; Kleinberg, L.; Li, M.; Florenes, V.A.; Wang, T.L.; Shih Ie, M. Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2006, 12, 5944–5950.
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327.
- Sato, H.; Takino, T.; Miyamori, H. Roles of membrane-type matrix metalloproteinase-1 in tumor invasion and metastasis. Cancer Sci. 2005, 96, 212–217.
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Mt1-mmp is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br. J. Cancer 2007, 97, 358–367.
- Zucker, S.; Hymowitz, M.; Conner, C.; Zarrabi, H.M.; Hurewitz, A.N.; Matrisian, L.; Boyd, D.; Nicolson, G.; Montana, S. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues. Clinical and experimental applications. Ann. N. Y. Acad. Sci. 1999, 878, 212–227.
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709.
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase mt1-mmp. J. Cell Biol. 2004, 167, 769–781.
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell Biol. 2009, 185, 11–19.
- Nyalendo, C.; Sartelet, H.; Gingras, D.; Beliveau, R. Inhibition of membrane-type 1 matrix metalloproteinase tyrosine phosphorylation blocks tumor progression in mice. Anticancer Res. 2010, 30, 1887–1895.
- Itoh, Y. Mt1-mmp: A key regulator of cell migration in tissue. IUBMB Life 2006, 58, 589–596.
- Galvez, B.G.; Matias-Roman, S.; Yanez-Mo, M.; Vicente-Manzanares, M.; Sanchez-Madrid, F.; Arroyo, A.G. Caveolae are a novel pathway for membrane-type 1 matrix metalloproteinase traffic in human endothelial cells. Mol. Biol. Cell 2004, 15, 678–687.
- Uekita, T.; Itoh, Y.; Yana, I.; Ohno, H.; Seiki, M. Cytoplasmic tail-dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity. J. Cell Biol. 2001, 155, 1345–1356.
- Wang, X.; Ma, D.; Keski-Oja, J.; Pei, D. Co-recycling of mt1-mmp and mt3-mmp through the trans-golgi network. Identification of dkv582 as a recycling signal. J. Biol. Chem. 2004, 279, 9331–9336.
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (mt1-mmp) and its vesicle-associated membrane protein 7 (vamp7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416.
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.K.; Chamness, G.C.; Elledge, R.M. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242.
- Nicholson, R.I.; Hutcheson, I.R.; Jones, H.E.; Hiscox, S.E.; Giles, M.; Taylor, K.M.; Gee, J.M. Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev. Endocr. Metab. Disord. 2007, 8, 241–253.
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622.
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112.
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009, 284, 12110–12124.
- Ceresa, B.P.; Bahr, S.J. Rab7 activity affects epidermal growth factor:Epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J. Biol. Chem. 2006, 281, 1099–1106.
- Wang, T.; Zhang, M.; Ma, Z.; Guo, K.; Tergaonkar, V.; Zeng, Q.; Hong, W. A role of rab7 in stabilizing egfr-her2 and in sustaining akt survival signal. J. Cell Physiol. 2012, 227, 2788–2797.
- Mizuno, K.; Kitamura, A.; Sasaki, T. Rabring7, a novel rab7 target protein with a ring finger motif. Mol. Biol. Cell 2003, 14, 3741–3752.
- Burger, A.M.; Gao, Y.; Amemiya, Y.; Kahn, H.J.; Kitching, R.; Yang, Y.; Sun, P.; Narod, S.A.; Hanna, W.M.; Seth, A.K. A novel ring-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005, 65, 10401–10412.
- Wymant, J.M.; Hiscox, S.; Westwell, A.D.; Urbe, S.; Clague, M.J.; Jones, A.T. The role of bca2 in the endocytic trafficking of egfr and significance as a prognostic biomarker in cancer. J. Cancer 2016, 7, 2388–2407.
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947.
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of pten. Biochem. J. 2012, 444, 457–464.
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of pkb/akt-dependent cell survival by the tumor suppressor pten. Cell 1998, 95, 29–39.
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. Pten functions by recruitment to cytoplasmic vesicles. Mol. Cell 2015, 58, 255–268.
- Shinde, S.R.; Maddika, S. Pten modulates egfr late endocytic trafficking and degradation by dephosphorylating rab7. Nat. Commun. 2016, 7, 10689.
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271.
- Davies, E.M.; Sheffield, D.A.; Tibarewal, P.; Fedele, C.G.; Mitchell, C.A.; Leslie, N.R. The pten and myotubularin phosphoinositide 3-phosphatases: Linking lipid signalling to human disease. Subcell. Biochem. 2012, 58, 281–336.
- Tibarewal, P.; Zilidis, G.; Spinelli, L.; Schurch, N.; Maccario, H.; Gray, A.; Perera, N.M.; Davidson, L.; Barton, G.J.; Leslie, N.R. Pten protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with akt activity. Sci. Signal. 2012, 5, ra18.
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Yan, C.; Zhao, T.; Du, H. Lysosomal acid lipase in cancer. Oncoscience 2015, 2, 727–728.
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of ppargamma in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2016, 7, 1529–1543.
- Zhao, T.; Du, H.; Ding, X.; Walls, K.; Yan, C. Activation of mtor pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(-/-) mice. Oncogene 2015, 34, 1938–1948.
- Ding, X.; Wu, L.; Yan, C.; Du, H. Establishment of lal-/- myeloid lineage cell line that resembles myeloid-derived suppressive cells. PLoS ONE 2015, 10, e0121001.
- Du, H.; Zhao, T.; Ding, X.; Yan, C. Hepatocyte-specific expression of human lysosome acid lipase corrects liver inflammation and tumor metastasis in lal(-/-) mice. Am. J. Pathol. 2015, 185, 2379–2389.
- Ding, X.; Du, H.; Yoder, M.C.; Yan, C. Critical role of the mtor pathway in development and function of myeloid-derived suppressor cells in lal-/- mice. Am. J. Pathol. 2014, 184, 397–408.
- Zhao, T.; Ding, X.; Du, H.; Yan, C. Myeloid-derived suppressor cells are involved in lysosomal acid lipase deficiency-induced endothelial cell dysfunctions. J. Immunol. 2014, 193, 1942–1953.
- Ding, X.; Zhang, W.; Zhao, T.; Yan, C.; Du, H. Rab7 gtpase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget 2017, 8, 30123–30137.
- Zhao, T.; Ding, X.; Yan, C.; Du, H. Endothelial rab7 gtpase mediates tumor growth and metastasis in lysosomal acid lipase-deficient mice. J. Biol. Chem. 2017, 292, 19198–19208.
- Edinger, A.L.; Cinalli, R.M.; Thompson, C.B. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Dev. Cell 2003, 5, 571–582.
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mtor-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288.
- Kan, O.; Baldwin, S.A.; Whetton, A.D. Apoptosis is regulated by the rate of glucose transport in an interleukin 3 dependent cell line. J. Exp. Med. 1994, 180, 917–923.
- Vander Heiden, M.G.; Plas, D.R.; Rathmell, J.C.; Fox, C.J.; Harris, M.H.; Thompson, C.B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell Biol. 2001, 21, 5899–5912.
- Whetton, A.D.; Bazill, G.W.; Dexter, T.M. Haemopoietic cell growth factor mediates cell survival via its action on glucose transport. EMBO J. 1984, 3, 409–413.
- Vander Heiden, M.G.; Chandel, N.S.; Schumacker, P.T.; Thompson, C.B. Bcl-xl prevents cell death following growth factor withdrawal by facilitating mitochondrial atp/adp exchange. Mol. Cell 1999, 3, 159–167.
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xl regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91, 627–637.
- Romero Rosales, K.; Peralta, E.R.; Guenther, G.G.; Wong, S.Y.; Edinger, A.L. Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis. Mol. Biol. Cell 2009, 20, 2831–2840.
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a role for the gtpase rab7 in prostate cancer progression. PLoS ONE 2014, 9, e87882.
- Steffan, J.J.; Cardelli, J.A. Thiazolidinediones induce rab7-rilp- mapk-dependent juxtanuclear lysosome aggregation and reduce tumor cell invasion. Traffic 2010, 11, 274–286.
- Cordonnier, M.N.; Dauzonne, D.; Louvard, D.; Coudrier, E. Actin filaments and myosin i alpha cooperate with microtubules for the movement of lysosomes. Mol. Biol. Cell 2001, 12, 4013–4029.
- Gupta, D.; Kono, T.; Evans-Molina, C. The role of peroxisome proliferator-activated receptor gamma in pancreatic beta cell function and survival: Therapeutic implications for the treatment of type 2 diabetes mellitus. Diabetes Obes. Metab. 2010, 12, 1036–1047.
- Kopelovich, L.; Fay, J.R.; Glazer, R.I.; Crowell, J.A. Peroxisome proliferator- activated receptor modulators as potential chemopreventive agents. Mol. Cancer Ther. 2002, 1, 357–363.
- Wang, W.; Zhang, H.; Liu, S.; Kim, C.K.; Xu, Y.; Hurley, L.A.; Nishikawa, R.; Nagane, M.; Hu, B.; Stegh, A.H.; et al. Internalized cd44s splice isoform attenuates egfr degradation by targeting rab7a. Proc. Natl. Acad. Sci. USA 2017, 114, 8366–8371.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Zhao, P.; Xu, Y.; Wei, Y.; Qiu, Q.; Chew, T.L.; Kang, Y.; Cheng, C. The cd44s splice isoform is a central mediator for invadopodia activity. J. Cell Sci. 2016, 129, 1355–1365.
- Laviolette, L.A.; Mermoud, J.; Calvo, I.A.; Olson, N.; Boukhali, M.; Steinlein, O.K.; Roider, E.; Sattler, E.C.; Huang, D.; Teh, B.T.; et al. Negative regulation of egfr signalling by the human folliculin tumour suppressor protein. Nat. Commun. 2017, 8, 15866.
- Birt, A.R.; Hogg, G.R.; Dube, W.J. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol. 1977, 113, 1674–1677.
- Okon, I.S.; Coughlan, K.A.; Zhang, C.; Moriasi, C.; Ding, Y.; Song, P.; Zhang, W.; Li, G.; Zou, M.H. Protein kinase lkb1 promotes rab7-mediated neuropilin-1 degradation to inhibit angiogenesis. J. Clin. Investig. 2014, 124, 4590–4602.
- Zachary, I.C. How neuropilin-1 regulates receptor tyrosine kinase signalling: The knowns and known unknowns. Biochem. Soc. Trans. 2011, 39, 1583–1591.
- Jia, H.; Cheng, L.; Tickner, M.; Bagherzadeh, A.; Selwood, D.; Zachary, I. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br. J. Cancer 2010, 102, 541–552.
- Oh, H.; Takagi, H.; Otani, A.; Koyama, S.; Kemmochi, S.; Uemura, A.; Honda, Y. Selective induction of neuropilin-1 by vascular endothelial growth factor (vegf): A mechanism contributing to vegf-induced angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 383–388.
- Gasparre, G.; Kurelac, I.; Capristo, M.; Iommarini, L.; Ghelli, A.; Ceccarelli, C.; Nicoletti, G.; Nanni, P.; De Giovanni, C.; Scotlandi, K.; et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene mtnd1, an oncojanus function. Cancer Res. 2011, 71, 6220–6229.
- Guerra, F.; Perrone, A.M.; Kurelac, I.; Santini, D.; Ceccarelli, C.; Cricca, M.; Zamagni, C.; De Iaco, P.; Gasparre, G. Mitochondrial DNA mutation in serous ovarian cancer: Implications for mitochondria-coded genes in chemoresistance. J. Clin. Oncol. 2012, 30, e373.
- Alonso-Curbelo, D.; Riveiro-Falkenbach, E.; Pérez-Guijarro, E.; Cifdaloz, M.; Karras, P.; Osterloh, L.; Megías, D.; Cañón, E.; Calvo, T.G.; Olmeda, D.; et al. Rab7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014, 26, 61–76.
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55.
- Alpaugh, M.L.; Tomlinson, J.S.; Shao, Z.M.; Barsky, S.H. A novel human xenograft model of inflammatory breast cancer. Cancer Res. 1999, 59, 5079–5084.
- Xiao, Y.; Ye, Y.; Yearsley, K.; Jones, S.; Barsky, S.H. The lymphovascular embolus of inflammatory breast cancer expresses a stem cell-like phenotype. Am. J. Pathol. 2008, 173, 561–574.
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890.
- Ye, Y.; Gao, J.X.; Tian, H.; Yearsley, K.; Lange, A.R.; Robertson, F.M.; Barsky, S.H. Early to intermediate steps of tumor embolic formation involve specific proteolytic processing of e-cadherin regulated by rab7. Mol. Cancer Res. 2012, 10, 713–726.
More
Information
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
2 times
(View History)
Update Date:
31 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No