RAB7 is a small guanosine triphosphatase (GTPase) extensively studied as regulator of vesicular trafficking. Indeed, its role is fundamental in several steps of the late endocytic pathway, including endosome maturation, transport from early endosomes to late endosomes and lysosomes, clustering and fusion of late endosomes and lysosomes in the perinuclear region and lysosomal biogenesis. Besides endocytosis, RAB7 is important for a number of other cellular processes among which, autophagy, apoptosis, signaling, and cell migration. Given the importance of RAB7 in these cellular processes, the interest to study the role of RAB7 in cancer progression is widely grown.
- RAB7
- endocytosis
- tumor progression
- cisplatin resistance
- extracellular vesicles
- oncogene
- oncosuppressor
1. Introduction
2. RAB7A in Cancer Progression
2.1. Oncogenic Functions of RAB7A
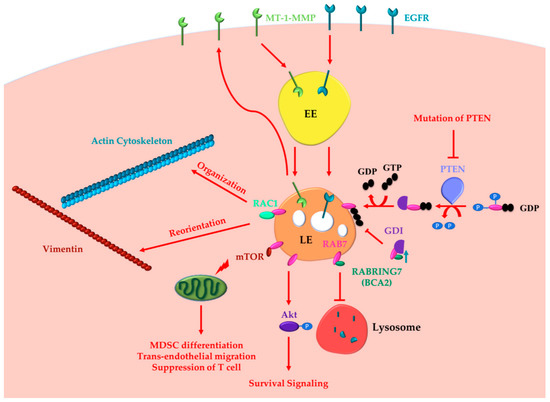
2.2. Oncosuppressor Functions of RAB7A
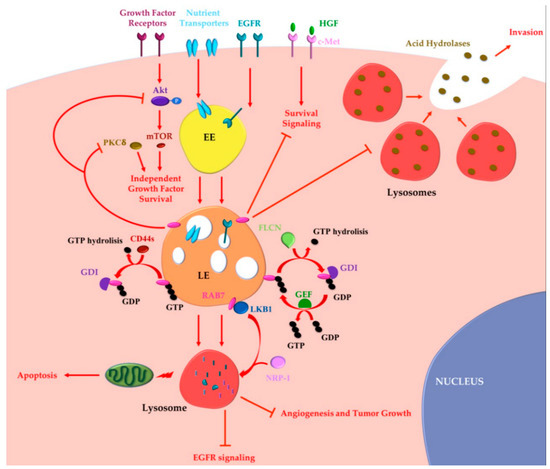
2.3. Is RAB7A an Oncojanus?
This entry is adapted from the peer-reviewed paper 10.3390/cancers11081096
References
- Pfeffer, S.R. Rab gtpases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715.
- Pfeffer, S.R. Rab gtpase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419.
- Zhen, Y.; Stenmark, H. Cellular functions of rab gtpases at a glance. J. Cell Sci. 2015, 128, 3171–3176.
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525.
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34.
- Langemeyer, L.; Frohlich, F.; Ungermann, C. Rab gtpase function in endosome and lysosome biogenesis. Trends Cell Biol. 2018, 28, 957–970.
- Kuchitsu, Y.; Fukuda, M. Revisiting rab7 functions in mammalian autophagy: Rab7 knockout studies. Cells 2018, 7, 215.
- Wen, H.; Zhan, L.; Chen, S.; Long, L.; Xu, E. Rab7 may be a novel therapeutic target for neurologic diseases as a key regulator in autophagy. J. Neurosci. Res. 2017, 95, 1993–2004.
- Snider, M.D. A role for rab7 gtpase in growth factor-regulated cell nutrition and apoptosis. Mol. Cell 2003, 12, 796–797.
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for rab7. Biochim. Biophys. Acta 2013, 1833, 503–510.
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small gtpase rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor trka. J. Neurosci. 2005, 25, 10930–10940.
- Deinhardt, K.; Salinas, S.; Verastegui, C.; Watson, R.; Worth, D.; Hanrahan, S.; Bucci, C.; Schiavo, G. Rab5 and rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 2006, 52, 293–305.
- Kawauchi, T.; Sekine, K.; Shikanai, M.; Chihama, K.; Tomita, K.; Kubo, K.; Nakajima, K.; Nabeshima, Y.; Hoshino, M. Rab gtpases-dependent endocytic pathways regulate neuronal migration and maturation through n-cadherin trafficking. Neuron 2010, 67, 588–602.
- Mottola, G. The complexity of rab5 to rab7 transition guarantees specificity of pathogen subversion mechanisms. Front. Cell Infect. Microbiol. 2014, 4, 180.
- Wozniak, A.L.; Long, A.; Jones-Jamtgaard, K.N.; Weinman, S.A. Hepatitis c virus promotes virion secretion through cleavage of the rab7 adaptor protein rilp. Proc. Natl. Acad. Sci. USA 2016, 113, 12484–12489.
- Croizet-Berger, K.; Daumerie, C.; Couvreur, M.; Courtoy, P.J.; van den Hove, M.F. The endocytic catalysts, rab5a and rab7, are tandem regulators of thyroid hormone production. Proc. Natl. Acad. Sci. USA 2002, 99, 8277–8282.
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of rab7a protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52.
- Cherfils, J.; Zeghouf, M. Regulation of small gtpases by gefs, gaps, and gdis. Physiol. Rev. 2013, 93, 269–309.
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. Gefs and gaps: Critical elements in the control of small g proteins. Cell 2007, 129, 865–877.
- Wang, T.; Ming, Z.; Xiaochun, W.; Hong, W. Rab7: Role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal. 2011, 23, 516–521.
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of rab7 mutant proteins associated with charcot-marie-tooth type 2b disease. J. Neurosci. 2008, 28, 1640–1648.
- De Luca, M.; Bucci, C. A new v-atpase regulatory mechanism mediated by the rab interacting lysosomal protein (rilp). Commun. Integr. Biol. 2014, 7, 1–4.
- Meggouh, F.; Bienfait, H.M.; Weterman, M.A.; de Visser, M.; Baas, F. Charcot-marie-tooth disease due to a de novo mutation of the rab7 gene. Neurology 2006, 67, 1476–1478.
- Houlden, H.; King, R.H.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel rab7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590.
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small gtp-ase late endosomal protein rab7 cause charcot-marie-tooth type 2b neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727.
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel rab7 mutation in a chinese family with charcot-marie-tooth type 2b disease. Gene 2014, 534, 431–434.
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799.
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction: A novel potential driver of epithelial-to-mesenchymal transition in cancer. Front. Oncol. 2017, 7, 295.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Mascia, A.; Gentile, F.; Izzo, A.; Mollo, N.; De Luca, M.; Bucci, C.; Nitsch, L.; Calì, G. Rab7 regulates cdh1 endocytosis, circular dorsal ruffles genesis and thyroglobulin internalization in a thyroid cell line. J. Cell Physiol. 2016, 231, 1695–1708.
- Carroll, B.; Mohd-Naim, N.; Maximiano, F.; Frasa, M.A.; McCormack, J.; Finelli, M.; Thoresen, S.B.; Perdios, L.; Daigaku, R.; Francis, R.E.; et al. The tbc/rabgap armus coordinates rac1 and rab7 functions during autophagy. Dev. Cell 2013, 25, 15–28.
- Frasa, M.A.; Maximiano, F.C.; Smolarczyk, K.; Francis, R.E.; Betson, M.E.; Lozano, E.; Goldenring, J.; Seabra, M.C.; Rak, A.; Ahmadian, M.R.; et al. Armus is a rac1 effector that inactivates rab7 and regulates e-cadherin degradation. Curr. Biol. 2010, 20, 198–208.
- Sun, Y.; Buki, K.G.; Ettala, O.; Vaaraniemi, J.P.; Vaananen, H.K. Possible role of direct rac1-rab7 interaction in ruffled border formation of osteoclasts. J. Biol. Chem. 2005, 280, 32356–32361.
- Herrmann, H.; Strelkov, S.V.; Burkhard, P.; Aebi, U. Intermediate filaments: Primary determinants of cell architecture and plasticity. J. Clin. Investig. 2009, 119, 1772–1783.
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771.
- Styers, M.L.; Kowalczyk, A.P.; Faundez, V. Intermediate filaments and vesicular membrane traffic: The odd couple’s first dance? Traffic 2005, 6, 359–365.
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883.
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851.
- Messica, Y.; Laser-Azogui, A.; Volberg, T.; Elisha, Y.; Lysakovskaia, K.; Eils, R.; Gladilin, E.; Geiger, B.; Beck, R. The role of vimentin in regulating cell invasive migration in dense cultures of breast carcinoma cells. Nano Lett. 2017, 17, 6941–6948.
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through rac1 and vimentin. Biochim. Biophys. Acta 2017, 1864, 367–381.
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small gtpase rab7a. Biochim. Biophys. Acta 2013, 1833, 1283–1293.
- Suwandittakul, N.; Reamtong, O.; Molee, P.; Maneewatchararangsri, S.; Sutherat, M.; Chaisri, U.; Wongkham, S.; Adisakwattana, P. Disruption of endocytic trafficking protein rab7 impairs invasiveness of cholangiocarcinoma cells. Cancer Biomark. 2017, 20, 255–266.
- Davidson, B.; Zhang, Z.; Kleinberg, L.; Li, M.; Florenes, V.A.; Wang, T.L.; Shih Ie, M. Gene expression signatures differentiate ovarian/peritoneal serous carcinoma from diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2006, 12, 5944–5950.
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327.
- Sato, H.; Takino, T.; Miyamori, H. Roles of membrane-type matrix metalloproteinase-1 in tumor invasion and metastasis. Cancer Sci. 2005, 96, 212–217.
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Mt1-mmp is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br. J. Cancer 2007, 97, 358–367.
- Zucker, S.; Hymowitz, M.; Conner, C.; Zarrabi, H.M.; Hurewitz, A.N.; Matrisian, L.; Boyd, D.; Nicolson, G.; Montana, S. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues. Clinical and experimental applications. Ann. N. Y. Acad. Sci. 1999, 878, 212–227.
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709.
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase mt1-mmp. J. Cell Biol. 2004, 167, 769–781.
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell Biol. 2009, 185, 11–19.
- Nyalendo, C.; Sartelet, H.; Gingras, D.; Beliveau, R. Inhibition of membrane-type 1 matrix metalloproteinase tyrosine phosphorylation blocks tumor progression in mice. Anticancer Res. 2010, 30, 1887–1895.
- Itoh, Y. Mt1-mmp: A key regulator of cell migration in tissue. IUBMB Life 2006, 58, 589–596.
- Galvez, B.G.; Matias-Roman, S.; Yanez-Mo, M.; Vicente-Manzanares, M.; Sanchez-Madrid, F.; Arroyo, A.G. Caveolae are a novel pathway for membrane-type 1 matrix metalloproteinase traffic in human endothelial cells. Mol. Biol. Cell 2004, 15, 678–687.
- Uekita, T.; Itoh, Y.; Yana, I.; Ohno, H.; Seiki, M. Cytoplasmic tail-dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity. J. Cell Biol. 2001, 155, 1345–1356.
- Wang, X.; Ma, D.; Keski-Oja, J.; Pei, D. Co-recycling of mt1-mmp and mt3-mmp through the trans-golgi network. Identification of dkv582 as a recycling signal. J. Biol. Chem. 2004, 279, 9331–9336.
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (mt1-mmp) and its vesicle-associated membrane protein 7 (vamp7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416.
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.K.; Chamness, G.C.; Elledge, R.M. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242.
- Nicholson, R.I.; Hutcheson, I.R.; Jones, H.E.; Hiscox, S.E.; Giles, M.; Taylor, K.M.; Gee, J.M. Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev. Endocr. Metab. Disord. 2007, 8, 241–253.
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622.
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112.
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009, 284, 12110–12124.
- Ceresa, B.P.; Bahr, S.J. Rab7 activity affects epidermal growth factor:Epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J. Biol. Chem. 2006, 281, 1099–1106.
- Wang, T.; Zhang, M.; Ma, Z.; Guo, K.; Tergaonkar, V.; Zeng, Q.; Hong, W. A role of rab7 in stabilizing egfr-her2 and in sustaining akt survival signal. J. Cell Physiol. 2012, 227, 2788–2797.
- Mizuno, K.; Kitamura, A.; Sasaki, T. Rabring7, a novel rab7 target protein with a ring finger motif. Mol. Biol. Cell 2003, 14, 3741–3752.
- Burger, A.M.; Gao, Y.; Amemiya, Y.; Kahn, H.J.; Kitching, R.; Yang, Y.; Sun, P.; Narod, S.A.; Hanna, W.M.; Seth, A.K. A novel ring-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005, 65, 10401–10412.
- Wymant, J.M.; Hiscox, S.; Westwell, A.D.; Urbe, S.; Clague, M.J.; Jones, A.T. The role of bca2 in the endocytic trafficking of egfr and significance as a prognostic biomarker in cancer. J. Cancer 2016, 7, 2388–2407.
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947.
- Zhang, X.C.; Piccini, A.; Myers, M.P.; Van Aelst, L.; Tonks, N.K. Functional analysis of the protein phosphatase activity of pten. Biochem. J. 2012, 444, 457–464.
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of pkb/akt-dependent cell survival by the tumor suppressor pten. Cell 1998, 95, 29–39.
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. Pten functions by recruitment to cytoplasmic vesicles. Mol. Cell 2015, 58, 255–268.
- Shinde, S.R.; Maddika, S. Pten modulates egfr late endocytic trafficking and degradation by dephosphorylating rab7. Nat. Commun. 2016, 7, 10689.
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271.
- Davies, E.M.; Sheffield, D.A.; Tibarewal, P.; Fedele, C.G.; Mitchell, C.A.; Leslie, N.R. The pten and myotubularin phosphoinositide 3-phosphatases: Linking lipid signalling to human disease. Subcell. Biochem. 2012, 58, 281–336.
- Tibarewal, P.; Zilidis, G.; Spinelli, L.; Schurch, N.; Maccario, H.; Gray, A.; Perera, N.M.; Davidson, L.; Barton, G.J.; Leslie, N.R. Pten protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with akt activity. Sci. Signal. 2012, 5, ra18.
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Yan, C.; Zhao, T.; Du, H. Lysosomal acid lipase in cancer. Oncoscience 2015, 2, 727–728.
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150.
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of ppargamma in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2016, 7, 1529–1543.
- Zhao, T.; Du, H.; Ding, X.; Walls, K.; Yan, C. Activation of mtor pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(-/-) mice. Oncogene 2015, 34, 1938–1948.
- Ding, X.; Wu, L.; Yan, C.; Du, H. Establishment of lal-/- myeloid lineage cell line that resembles myeloid-derived suppressive cells. PLoS ONE 2015, 10, e0121001.
- Du, H.; Zhao, T.; Ding, X.; Yan, C. Hepatocyte-specific expression of human lysosome acid lipase corrects liver inflammation and tumor metastasis in lal(-/-) mice. Am. J. Pathol. 2015, 185, 2379–2389.
- Ding, X.; Du, H.; Yoder, M.C.; Yan, C. Critical role of the mtor pathway in development and function of myeloid-derived suppressor cells in lal-/- mice. Am. J. Pathol. 2014, 184, 397–408.
- Zhao, T.; Ding, X.; Du, H.; Yan, C. Myeloid-derived suppressor cells are involved in lysosomal acid lipase deficiency-induced endothelial cell dysfunctions. J. Immunol. 2014, 193, 1942–1953.
- Ding, X.; Zhang, W.; Zhao, T.; Yan, C.; Du, H. Rab7 gtpase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget 2017, 8, 30123–30137.
- Zhao, T.; Ding, X.; Yan, C.; Du, H. Endothelial rab7 gtpase mediates tumor growth and metastasis in lysosomal acid lipase-deficient mice. J. Biol. Chem. 2017, 292, 19198–19208.
- Edinger, A.L.; Cinalli, R.M.; Thompson, C.B. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Dev. Cell 2003, 5, 571–582.
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mtor-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288.
- Kan, O.; Baldwin, S.A.; Whetton, A.D. Apoptosis is regulated by the rate of glucose transport in an interleukin 3 dependent cell line. J. Exp. Med. 1994, 180, 917–923.
- Vander Heiden, M.G.; Plas, D.R.; Rathmell, J.C.; Fox, C.J.; Harris, M.H.; Thompson, C.B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell Biol. 2001, 21, 5899–5912.
- Whetton, A.D.; Bazill, G.W.; Dexter, T.M. Haemopoietic cell growth factor mediates cell survival via its action on glucose transport. EMBO J. 1984, 3, 409–413.
- Vander Heiden, M.G.; Chandel, N.S.; Schumacker, P.T.; Thompson, C.B. Bcl-xl prevents cell death following growth factor withdrawal by facilitating mitochondrial atp/adp exchange. Mol. Cell 1999, 3, 159–167.
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xl regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91, 627–637.
- Romero Rosales, K.; Peralta, E.R.; Guenther, G.G.; Wong, S.Y.; Edinger, A.L. Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis. Mol. Biol. Cell 2009, 20, 2831–2840.
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a role for the gtpase rab7 in prostate cancer progression. PLoS ONE 2014, 9, e87882.
- Steffan, J.J.; Cardelli, J.A. Thiazolidinediones induce rab7-rilp- mapk-dependent juxtanuclear lysosome aggregation and reduce tumor cell invasion. Traffic 2010, 11, 274–286.
- Cordonnier, M.N.; Dauzonne, D.; Louvard, D.; Coudrier, E. Actin filaments and myosin i alpha cooperate with microtubules for the movement of lysosomes. Mol. Biol. Cell 2001, 12, 4013–4029.
- Gupta, D.; Kono, T.; Evans-Molina, C. The role of peroxisome proliferator-activated receptor gamma in pancreatic beta cell function and survival: Therapeutic implications for the treatment of type 2 diabetes mellitus. Diabetes Obes. Metab. 2010, 12, 1036–1047.
- Kopelovich, L.; Fay, J.R.; Glazer, R.I.; Crowell, J.A. Peroxisome proliferator- activated receptor modulators as potential chemopreventive agents. Mol. Cancer Ther. 2002, 1, 357–363.
- Wang, W.; Zhang, H.; Liu, S.; Kim, C.K.; Xu, Y.; Hurley, L.A.; Nishikawa, R.; Nagane, M.; Hu, B.; Stegh, A.H.; et al. Internalized cd44s splice isoform attenuates egfr degradation by targeting rab7a. Proc. Natl. Acad. Sci. USA 2017, 114, 8366–8371.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Zhao, P.; Xu, Y.; Wei, Y.; Qiu, Q.; Chew, T.L.; Kang, Y.; Cheng, C. The cd44s splice isoform is a central mediator for invadopodia activity. J. Cell Sci. 2016, 129, 1355–1365.
- Laviolette, L.A.; Mermoud, J.; Calvo, I.A.; Olson, N.; Boukhali, M.; Steinlein, O.K.; Roider, E.; Sattler, E.C.; Huang, D.; Teh, B.T.; et al. Negative regulation of egfr signalling by the human folliculin tumour suppressor protein. Nat. Commun. 2017, 8, 15866.
- Birt, A.R.; Hogg, G.R.; Dube, W.J. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol. 1977, 113, 1674–1677.
- Okon, I.S.; Coughlan, K.A.; Zhang, C.; Moriasi, C.; Ding, Y.; Song, P.; Zhang, W.; Li, G.; Zou, M.H. Protein kinase lkb1 promotes rab7-mediated neuropilin-1 degradation to inhibit angiogenesis. J. Clin. Investig. 2014, 124, 4590–4602.
- Zachary, I.C. How neuropilin-1 regulates receptor tyrosine kinase signalling: The knowns and known unknowns. Biochem. Soc. Trans. 2011, 39, 1583–1591.
- Jia, H.; Cheng, L.; Tickner, M.; Bagherzadeh, A.; Selwood, D.; Zachary, I. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br. J. Cancer 2010, 102, 541–552.
- Oh, H.; Takagi, H.; Otani, A.; Koyama, S.; Kemmochi, S.; Uemura, A.; Honda, Y. Selective induction of neuropilin-1 by vascular endothelial growth factor (vegf): A mechanism contributing to vegf-induced angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 383–388.
- Gasparre, G.; Kurelac, I.; Capristo, M.; Iommarini, L.; Ghelli, A.; Ceccarelli, C.; Nicoletti, G.; Nanni, P.; De Giovanni, C.; Scotlandi, K.; et al. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene mtnd1, an oncojanus function. Cancer Res. 2011, 71, 6220–6229.
- Guerra, F.; Perrone, A.M.; Kurelac, I.; Santini, D.; Ceccarelli, C.; Cricca, M.; Zamagni, C.; De Iaco, P.; Gasparre, G. Mitochondrial DNA mutation in serous ovarian cancer: Implications for mitochondria-coded genes in chemoresistance. J. Clin. Oncol. 2012, 30, e373.
- Alonso-Curbelo, D.; Riveiro-Falkenbach, E.; Pérez-Guijarro, E.; Cifdaloz, M.; Karras, P.; Osterloh, L.; Megías, D.; Cañón, E.; Calvo, T.G.; Olmeda, D.; et al. Rab7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014, 26, 61–76.
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55.
- Alpaugh, M.L.; Tomlinson, J.S.; Shao, Z.M.; Barsky, S.H. A novel human xenograft model of inflammatory breast cancer. Cancer Res. 1999, 59, 5079–5084.
- Xiao, Y.; Ye, Y.; Yearsley, K.; Jones, S.; Barsky, S.H. The lymphovascular embolus of inflammatory breast cancer expresses a stem cell-like phenotype. Am. J. Pathol. 2008, 173, 561–574.
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890.
- Ye, Y.; Gao, J.X.; Tian, H.; Yearsley, K.; Lange, A.R.; Robertson, F.M.; Barsky, S.H. Early to intermediate steps of tumor embolic formation involve specific proteolytic processing of e-cadherin regulated by rab7. Mol. Cancer Res. 2012, 10, 713–726.