 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christina Wasmus | + 2436 word(s) | 2436 | 2021-07-09 09:56:05 | | | |
| 2 | Nora Tang | Meta information modification | 2436 | 2021-08-20 10:13:08 | | |
Video Upload Options
The heart is the most energy-consuming organ in the human body. In heart failure, the homeostasis of energy supply and demand is endangered by an increase in cardiomyocyte workload, or by an insufficiency in energy-providing processes. Energy metabolism is directly associated with mitochondrial redox homeostasis. The production of toxic reactive oxygen species (ROS) may overwhelm mitochondrial and cellular ROS defense mechanisms in case of heart failure. Mitochondria are essential cell organelles and provide 95% of the required energy in the heart. Metabolic remodeling, changes in mitochondrial structure or function, and alterations in mitochondrial calcium signaling diminish mitochondrial energy provision in many forms of cardiomyopathy. The mitochondrial respiratory chain creates a proton gradient across the inner mitochondrial membrane, which couples respiration with oxidative phosphorylation and the preservation of energy in the chemical bonds of ATP. Akin to other mitochondrial enzymes, the respiratory chain is integrated into the inner mitochondrial membrane. The tight association with the mitochondrial phospholipid cardiolipin (CL) ensures its structural integrity and coordinates enzymatic activity. This review focuses on how changes in mitochondrial CL may be associated with heart failure. Dysfunctional CL has been found in diabetic cardiomyopathy, ischemia reperfusion injury and the aging heart. Barth syndrome (BTHS) is caused by an inherited defect in the biosynthesis of cardiolipin. Moreover, a dysfunctional CL pool causes other types of rare inherited cardiomyopathies, such as Sengers syndrome and Dilated Cardiomyopathy with Ataxia (DCMA). Here we review the impact of cardiolipin deficiency on mitochondrial functions in cellular and animal models.
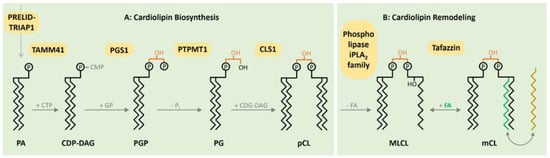
1. CL Biosynthesis
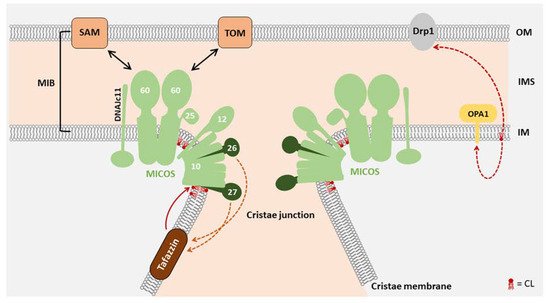
2. Function of CL in Mitochondrial Morphology
| Function | Anmial | Human |
|---|---|---|
| Mitochondrial morphology | A tafazzin knockdown mouse model of Barth syndrome describes alteration of mitochondrial phospholipid compositions via lipidomics [23]. Morphology alterations as mitochondrial enlargement, concentric layers of cristae or large vacuoles were observed in tafazzin-deficient mice [24]. |
BTHS patient-derived lymphoblasts (BTHS lymphoblasts) reveal enlarged mitochondria with a lower surface area of cristae and altered morphology [25]. Another study with BTHS lymphoblasts detected giant, partly onion-shaped mitochondria [26]. |
| Oxidative phosphorylation | A BTHS mouse model with an inducible systemic knockdown of tafazzin gene shows a reduced respiration on succinate as well as on pyruvate and malate. Furthermore, respirasome remodeling was detected [27]. | BTHS patient-derived induced pluripotent stem cells (BTHS-iPSC) reveal structural remodeling of respiratory chain complexes resulting in decreased mitochondrial respiration [28]. In a second study, mitochondria of BTHS lymphoblasts show a reduced respiratory activity on succinate and ascorbate [25]. |
| Krebs cycle | BTHS mouse model reveals a striking reduction in succinate dehydrogenase activity in cardiac mitochondria [27]. | BTHS skin fibroblasts reveal a significant destabilization of 2-oxoglutarate dehydrogenase and branched-chain ketoacid dehydrogenase [29]. |
| Apoptosis | Murine germline TAZ knockout mice model reveals significant increased cardiomyocyte apoptosis and fibrosis occurrence [30]. | BTHS lymphoblasts show a requirement of cardiolipin for apoptosis and an apoptotic defect [31]. |
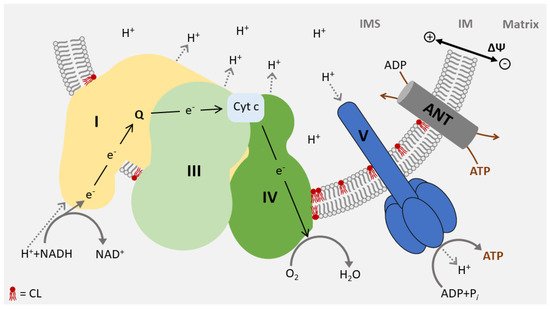
3. Function of CL in Energy Metabolism
References
- Schlame, M. Protein crowding in the inner mitochondrial membrane. Biochim. Biophys. Acta Bioenerg. 2020, 1862, 148305.
- Gebert, N.; Joshi, A.S.; Kutik, S.; Becker, T.; McKenzie, M.; Guan, X.L.; Mooga, V.P.; Stroud, D.A.; Kulkarni, G.; Wenk, M.R.; et al. Mitochondrial Cardiolipin Involved in Outer-Membrane Protein Biogenesis: Implications for Barth Syndrome. Curr. Biol. 2009, 19, 2133–2139.
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291.
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial Phosphatase PTPMT1 Is Essential for Cardiolipin Biosynthesis. Cell Metab. 2011, 13, 690–700.
- Kasahara, T.; Kubota-Sakashita, M.; Nagatsuka, Y.; Hirabayashi, Y.; Hanasaka, T.; Tohyama, K.; Kato, T. Cardiolipin is essential for early embryonic viability and mitochondrial integrity of neurons in mammals. FASEB J. 2019, 34, 1465–1480.
- Dinca, A.A.; Chien, W.-M.; Chin, M.T. Identification of novel mitochondrial localization signals in human Tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome. J. Mol. Cell. Cardiol. 2018, 114, 83–92.
- Hijikata, A.; Yura, K.; Ohara, O.; Go, M. Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain. Meta Gene 2015, 4, 92–106.
- Xu, Y.; Zhang, S.; Malhotra, A.; Edelman-Novemsky, I.; Ma, J.; Kruppa, A.; Cernicica, C.; Blais, S.; Neubert, T.A.; Ren, M.; et al. Characterization of Tafazzin Splice Variants from Humans and Fruit Flies. J. Biol. Chem. 2009, 284, 29230–29239.
- Schlame, M.; Xu, Y.; Ren, M. The Basis for Acyl Specificity in the Tafazzin Reaction. J. Biol. Chem. 2017, 292, 5499–5506.
- Xu, Y.; Anjaneyulu, M.; Donelian, A.; Yu, W.; Greenberg, M.L.; Ren, M.; Owusu-Ansah, E.; Schlame, M. Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc. Natl. Acad. Sci. USA 2019, 116, 11235–11240.
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-coenzyme A Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371.
- Mejia, E.M.; Zegallai, H.; Bouchard, E.D.; Banerji, V.; Ravandi, A.; Hatch, G.M. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J. Biol. Chem. 2018, 293, 7564–7577.
- Miklas, J.W.; Clark, E.; Levy, S.; Detraux, D.; Leonard, A.; Beussman, K.; Showalter, M.R.; Smith, A.T.; Hofsteen, P.; Yang, X.; et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 2019, 10, 1–21.
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165.
- Baandrup, U.; Florio, R.A.; Roters, F.; Olsen, E.G. Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy. A semiquantitative study in 48 patients. Circulation 1981, 63, 1289–1298.
- Sabbah, H.N.; Sharov, V.; Riddle, J.M.; Kono, T.; Lesch, M.; Goldstein, S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 1992, 24, 1333–1347.
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 3–7.
- Frohman, M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. 2015, 93, 263–269.
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99.
- Sharov, V.G.; Sabbah, H.N.; Shimoyama, H.; Goussev, A.V.; Lesch, M.; Goldstein, S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am. J. Pathol. 1996, 148, 141–149.
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803.
- Meglei, G.; McQuibban, G.A. The Dynamin-Related Protein Mgm1p Assembles into Oligomers and Hydrolyzes GTP To Function in Mitochondrial Membrane Fusion†. Biochemistry 2009, 48, 1774–1784.
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325.
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and Skeletal Muscle Defects in a Mouse Model of Human Barth Syndrome. J. Biol. Chem. 2010, 286, 899–908.
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.-P.; Landes, T.; Arnauné-Pelloquin, L.; Vial, G.; Taleux, N.; Slomianny, C.; et al. Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1194–1206.
- Acehan, D.; Xu, Y.; Stokes, D.L.; Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Investig. 2007, 87, 40–48.
- Dudek, J.; Cheng, I.F.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med 2016, 8, 139–154.
- Dudek, J.; Cheng, I.-F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.J.A.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819.
- Chatzispyrou, I.A.; Guerrero-Castillo, S.; Held, N.M.; Ruiter, J.P.; Denis, S.W.; Ijlst, L.; Wanders, R.J.; Van Weeghel, M.; Ferdinandusse, S.; Vaz, F.M.; et al. Barth syndrome cells display widespread remodeling of mitochondrial complexes without affecting metabolic flux distribution. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3650–3658.
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 2020, 126, 1024–1039.
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.A.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008, 183, 681–696.
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; Van Der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056.
- Rampelt, H.; Zerbes, R.M.; Van Der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 737–746.
- Weber, T.A.; Koob, S.; Heide, H.; Wittig, I.; Head, B.; Van Der Bliek, A.; Brandt, U.; Mittelbronn, M.; Reichert, A.S. APOOL Is a Cardiolipin-Binding Constituent of the Mitofilin/MINOS Protein Complex Determining Cristae Morphology in Mammalian Mitochondria. PLoS ONE 2013, 8, e63683.
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J.; Youle, R.J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 2015, 4, e07739.
- Koob, S.; Barrera, M.; Anand, R.; Reichert, A.S. The non-glycosylated isoform of MIC26 is a constituent of the mammalian MICOS complex and promotes formation of crista junctions. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 1551–1563.
- Van Strien, J.; Guerrero-Castillo, S.; Chatzispyrou, I.A.; Houtkooper, R.H.; Brandt, U.; Huynen, M.A. COmplexome Profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics 2019, 35, 3083–3091.
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328.
- Anand, R.; Kondadi, A.K.; Meisterknecht, J.; Golombek, M.; Nortmann, O.; Riedel, J.; Peifer-Weiß, L.; Brocke-Ahmadinejad, N.; Schlütermann, D.; Stork, B.; et al. MIC26 and MIC27 cooperate to regulate cardiolipin levels and the landscape of OXPHOS complexes. Life Sci. Alliance 2020, 3, e202000711.
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018, 28, 67–76.
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature 2016, 538, 406–410.
- Sedlák, E.; Robinson, N.C. Phospholipase A2 Digestion of Cardiolipin Bound to Bovine CytochromecOxidase Alters Both Activity and Quaternary Structure†. Biochemistry 1999, 38, 14966–14972.
- Sedlák, E.; Panda, M.; Dale, M.P.; Weintraub, S.T.; Robinson, N.C. Photolabeling of Cardiolipin Binding Subunits within Bovine Heart CytochromecOxidase†. Biochemistry 2006, 45, 746–754.
- Shinzawa-Itoh, K.; Seiyama, J.; Terada, H.; Nakatsubo, R.; Naoki, K.; Nakashima, Y.; Yoshikawa, S. Bovine Heart NADH−Ubiquinone Oxidoreductase Contains One Molecule of Ubiquinone with Ten Isoprene Units as One of the Cofactors. Biochemistry 2010, 49, 487–492.
- Sharpley, M.S.; Shannon, R.J.; Draghi, A.F.; Hirst, J. Interactions between Phospholipids and NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Mitochondria†. Biochemistry 2006, 45, 241–248.
- Robinson, N.C.; Zborowski, J.; Talbert, L.H. Cardiolipin-depleted bovine heart cytochrome c oxidase: Binding stoichiometry and affinity for cardiolipin derivatives. Biochemistry 1990, 29, 8962–8969.
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526.
- Laird, D.M.; Eble, K.S.; Cunningham, C.C. Reconstitution of mitochondrial F0.F1-ATPase with phosphatidylcholine using the nonionic detergent, octylglucoside. J. Biol. Chem. 1986, 261, 14844–14850.
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692.
- Mileykovskaya, E.; Penczek, P.A.; Fang, J.; Mallampalli, V.K.P.S.; Sparagna, G.C.; Dowhan, W. Arrangement of the Respiratory Chain Complexes in Saccharomyces cerevisiae Supercomplex III2IV2 Revealed by Single Particle Cryo-Electron Microscopy. J. Biol. Chem. 2012, 287, 23095–23103.
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880.
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin Is Essential for Organization of Complexes III and IV into a Supercomplex in Intact Yeast Mitochondria. J. Biol. Chem. 2005, 280, 29403–29408.
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of Cardiolipin in thecrd1Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function. J. Biol. Chem. 2000, 275, 22387–22394.
- Gómez, L.A.; Hagen, T.M. Age-related decline in mitochondrial bioenergetics: Does supercomplex destabilization determine lower oxidative capacity and higher superoxide production? Semin. Cell Dev. Biol. 2012, 23, 758–767.
- Ostrander, D.B.; Sparagna, G.C.; Amoscato, A.A.; McMillin, J.B.; Dowhan, W. Decreased Cardiolipin Synthesis Corresponds with Cytochromec Release in Palmitate-induced Cardiomyocyte Apoptosis. J. Biol. Chem. 2001, 276, 38061–38067.
- Gadicherla, A.K.; Stowe, D.F.; Antholine, W.E.; Yang, M.; Camara, A.K. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 419–429.
- Saini-Chohan, H.K.; Holmes, M.G.; Chicco, A.J.; Taylor, W.A.; Moore, R.L.; McCune, S.A.; Hickson-Bick, D.L.; Hatch, G.M.; Sparagna, G.C. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 2008, 50, 1600–1608.
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.V.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484.
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. eLife 2020, 9.
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886.
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; et al. Essential Role for Mitochondrial Thioredoxin Reductase in Hematopoiesis, Heart Development, and Heart Function. Mol. Cell. Biol. 2004, 24, 9414–9423.
- Ho, Y.-S.; Magnenat, J.-L.; Gargano, M.; Cao, J. The Nature of Antioxidant Defense Mechanisms: A Lesson from Transgenic Studies. Environ. Health Perspect. 1998, 106, 1219.
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; Clair, D.K.S. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996, 98, 1253–1260.


