1. CL Biosynthesis
Mitochondrial membranes are characterized by their high content of membrane proteins and unique phospholipid composition. A recent study found the respiratory chain, the ADP/ATP carrier, phosphatidylethanolamine, phosphatidylcholine and cardiolipin to be the main constituents of the inner membrane
[1][10]. The hallmark lipid is the dimeric phospholipid cardiolipin (CL), which is found almost exclusively in the inner mitochondrial membrane. Four different fatty acids can be bound to CL forming different CL species, leading to a highly diversified CL pool
[2][11]. CL in the colon has short and mostly saturated fatty acids, whereas CL in the brain primarily constitutes long and unsaturated CL species
[3][12]. The heart has an unique CL pool consisting mostly of tetralinoleoyl-CL (CL(18:2)). Interestingly, the highest proportions of oxidized CL were found in heart (1.8% ± 0.7%) and skeletal muscle
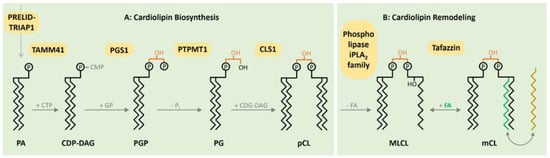
[3][12]. The diversified CL pool is acquired by reshaping CL acyl composition after its initial biosynthesis. CL is synthesized by enzymes located in the inner mitochondrial membrane. An important step in the biosynthesis of CL in the inner membrane is catalyzed by PTPMT1, converting phosphatidylglycerol phosphate to phosphatidylglycerol (
Figure 1). Deletion of this in mice is embryonically lethal and PTPMT1-deficient Mouse Embryonal Fibroblasts (MEFs) of embryos obtained from intercrosses of PTPMT1+/flox mice reveal the essential role of this enzyme in CL biosynthesis and affect mitochondrial morphology and respiration
[4][13]. CL Synthase (CLS1) catalyzes the subsequent addition of a second molecule of CDP-DAG to form premature CL (
Figure 1). The embryonic lethality of CLS1-deficient animals underscores the essential role of CLS1 in CL biosynthesis. Neuron-specific knockout results in neuronal loss and gliosis in the forebrains as a result of defective respiration and morphological abnormalities in these mitochondria
[5][14].
Figure 1. Biosynthesis and remodeling of cardiolipin (CL) in the inner membrane. Phosphatic acid (PA) as a precursor reaches the mitochondrial matrix via the PRELID–TRIAP1 complex. The CDP-DAG synthase TAMM41 activates PA with cytidine triphosphate (CTP), producing CDP-DAG. Subsequently, the phosphatidylglycerol phosphate synthase 1 (PGS1) catalyzes the formation of phosphatidylglycerol phosphate (PGP), followed by the dephosphorylation generating phosphatidylglycerol (PG) via the mitochondrial protein-tyrosine phosphatase 1 (PTPMT1). In a last reaction, Cardiolipin synthase 1 (CLS1) mediates the emergence of premature CL based on one molecule of PG and one CDG-DAG. Following the initial synthesis, CL remodeling ensues. After the removement of fatty acids (FAs) mediated by a member of the phospholipase iPLA2 family, the incorporation of new FAs is proceeded by Tafazzin to form matured CL from Monolysocardiolipn (MLCL). To reach an energetic optimum, the remodeling and the ongoing exchange of FAs is a continual persisting process, culminating in a highly diversified CL pool.
Afterwards, CL remodeling is initiated by deacylation, catalyzed by mitochondrial members of the Ca2+ independent phospholipases to form monolysocardiolipin (MLCL). Subsequently, CL is reacetylated by Tafazzin to form mature CL (Figure 1).
The protein Tafazzin is produced by alternative splicing of the TAZ gene. The gene consists of 11 exons and two ATG initiation sites. Transcription of the TAZ gene gives rise to multiple mRNAs by alternative splicing at exons 5–7. A highly hydrophobic segment of 30 residues at the
N-terminus acts as a membrane anchor. Tafazzin is directed to mitochondria with the help of two independent targeting sequences
[6][15]. The active site of phospholipid-binding has been identified in a 57-amino acid cleft containing positively charged residues
[7][16]. The substrate specificity of Tafazzin was found to be surprisingly low and cannot explain the tissue-specific remodeling of the CL pool
[8][17]. Recent data suggest that Tafazzin’s substrate specificity is determined by the thermodynamic properties of lipid domains in the vicinity of the enzyme
[8][17]. Tafazzin exchanges fatty acids in the CL molecule until the molecular species composition with optimal packing conditions and the lowest free energy is established
[9][18]. A recent study from the Schlame lab suggests that the thermodynamic properties of lipid domains are determined by the assembly of respiratory chain complexes. This study proposes that protein crowding in the respiratory chain imposes packing stress on the lipid bilayer, which is relieved by CL remodeling to form tightly packed lipid–protein complexes
[10][19]. Further evidence was provided by an integrative approach of lipidomics and transcriptomics. Oemer et al. found that a transcriptional regulation of CL biosynthesis genes shows no correlation with the tissue-specific CL composition. Interestingly, the only significant compliance with CL content was found for genes of the respiratory chain. This finding supports the idea that the respiratory chain and other protein complexes determine the tissue-specific CL composition of the membrane and hence its properties.
Besides Tafazzin, two other enzymes are involved in CL remodeling. The Coenzyme A-dependent monolysocardiolipin acyltransferase (MLCLAT1) is a splice variant of the HADHA gene encoding for the α-subunit of the human trifunctional enzyme (α-TFP)
[11][12][20,21]. In a recent study, Miklas et al. show that apart from its function in the β-oxidation cycle, α-TFP plays a role in CL remodeling
[13][22]. As patient-derived HADHA knockout cardiomyocytes do not accumulate MLCL as TAZ mutant cells, it has been concluded that HADHA acts subsequently to TAZ to mature the CL pool. The second enzyme is the ER-MAM resident lysocardiolipin acyltransferase (ALCAT1). Overexpression of ALCAT1 in mouse myoblasts increases the incorporation of docosahexaenoic acid (22:6) into CL, yielding a peroxidation prone form of CL, which has been suggested to reflect a pathogenic CL remodeling
[14][23].
2. Function of CL in Mitochondrial Morphology
Structural abnormalities of mitochondria, including fragmentation, disruption of mitochondrial membranes and loss of the electron-dense matrix have been observed in animal models of heart failure and in patients suffering from hypertrophic cardiomyopathy
[15][24]. In a dog model of chronic heart failure, the mitochondrial ultrastructure changes from tightly packed to disorganized cristae
[16][25]. Mitochondria possess two highly specialized membranes—the outer and the inner mitochondrial membrane. Invaginations in the inner membrane form the cristae structures, which are areas of intensive membrane curvature. CL plays a considerable role in shaping the morphology of mitochondria and prominently locates in the bended regions of the inner membrane
[17][26]. CL adopts a cone-shaped structure due to the high content of unsaturated fatty acids, which is causative for the membrane bending at sites of high CL concentration. In addition, mitochondrial morphology is also shaped by a constant remodeling through the fission and fusion of individual mitochondria
[18][27]. The opposing processes of fusion (merging of two mitochondria) and fission (segregation of two mitochondria) are shown to form a highly dynamic network of mitochondria in the cell. The continuing remodeling by fission and fusion is essential for normal mitochondrial function, downregulation of mitochondrial fusion promotes apoptosis and cardiomyocyte loss
[19][28]. Mitochondrial fragmentation was elucidated to influence heart failure notably
[20][29]. By its role in regulating mitochondrial fission and fusion, CL actively participates in shaping the mitochondrial network. Fission and fusion are regulated by a set of dynamin-related GTPases, whereby CL serves as a modulator of the activity of the fission protein Drp1 and the fusion protein OPA1
[21][22][30,31]. Morphological alterations of mitochondria are evident in human patients with defects in CL biosynthesis and remodeling as well as in animal models (
Table 1).
Table 1. Role of CL in inherited cardiomyopathies in animal models and human.
| Function |
Anmial |
Human |
| Mitochondrial morphology |
A tafazzin knockdown mouse model of Barth syndrome describes alteration of mitochondrial phospholipid compositions via lipidomics [23][32].
Morphology alterations as mitochondrial enlargement, concentric layers of cristae or large vacuoles were observed in tafazzin-deficient mice [24][33]. |
BTHS patient-derived lymphoblasts (BTHS lymphoblasts) reveal enlarged mitochondria with a lower surface area of cristae and altered morphology [25][34].
Another study with BTHS lymphoblasts detected giant, partly onion-shaped mitochondria [26][35]. |
| Oxidative phosphorylation |
A BTHS mouse model with an inducible systemic knockdown of tafazzin gene shows a reduced respiration on succinate as well as on pyruvate and malate. Furthermore, respirasome remodeling was detected [27][36]. |
BTHS patient-derived induced pluripotent stem cells (BTHS-iPSC) reveal structural remodeling of respiratory chain complexes resulting in decreased mitochondrial respiration [28][37].
In a second study, mitochondria of BTHS lymphoblasts show a reduced respiratory activity on succinate and ascorbate [25][34]. |
| Krebs cycle |
BTHS mouse model reveals a striking reduction in succinate dehydrogenase activity in cardiac mitochondria [27][36]. |
BTHS skin fibroblasts reveal a significant destabilization of 2-oxoglutarate dehydrogenase and branched-chain ketoacid dehydrogenase [29][38]. |
| Apoptosis |
Murine germline TAZ knockout mice model reveals significant increased cardiomyocyte apoptosis and fibrosis occurrence [30][39]. |
BTHS lymphoblasts show a requirement of cardiolipin for apoptosis and an apoptotic defect [31][40]. |
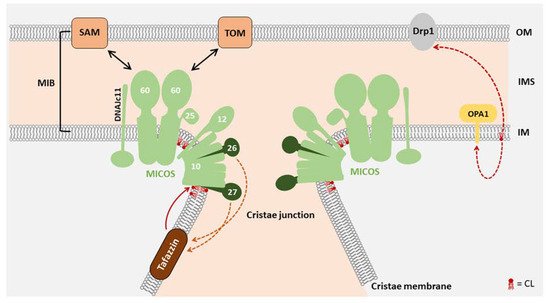
The formation of cristae structures critically depends on the mitochondrial contact site and cristae organizing system (MICOS). These protein complexes locate at the cristae junctions and seal individual cristae to maintain individual membrane potentials
[32][8]. The MICOS complex is a central component of a large interaction network which includes several complexes in the inner membrane and outer membrane
[33][41]. MIC27 (APOOL) as a MICOS complex constituent directly interacts with CL and this interaction was found to be essential for MIC27 assembly in the MICOS complex
[34][35][42,43] (
Figure 2). MIC27 and the structurally related MIC26 also modulate the cardiolipin remodeling enzyme Tafazzin (see below)
[36][44]. In fibroblasts from Barth syndrome patients, a compensatory increase in MICOS subunits was observed
[29][37][38,45]. Interestingly, a slightly lower molecular mass was detected for MICOS subunits in Barth syndrome (BTHS) fibroblasts, indicative of structural changes due to CL deficiency. Application of the CL-interacting molecule SS31/Elamipretide normalized deregulated structural proteins, such as Drp1, Mfn2, Opa1 and Mic60 in human heart failure patients
[38][46].
Figure 2. The role of CL in mitochondrial morphology: The mitochondrial contact site and cristae organizing system (MICOS) complex requires CL for optimal structural integrity. MICOS is a protein complex located in the inner mitochondrial membrane, playing an essential role in cristae junction formation. The resulting membrane invaginations harbor the respiratory chain complexes. By interactions with other proteins in the outer mitochondrial membrane such as the Translocase of the Outer membrane (TOM) and the sorting and Assembly Machinery (SAM), the mitochondrial intermembrane space (IMS) bridging complex (MIB) is formed. The MICOS subunits MIC27 and MIC26 influence the regulation of CL levels, while CL is remodeled by Tafazzin
[39][47]. Not only the integration of MIC27 in MICOS, but also the function of other mitochondrial membrane anchored proteins such as the fission and fusion proteins Drp1 and OPA1 is affected by CL
[40][48]. OM, outer membrane; IMS, intermembrane space; IM, inner membrane.
3. Function of CL in Energy Metabolism
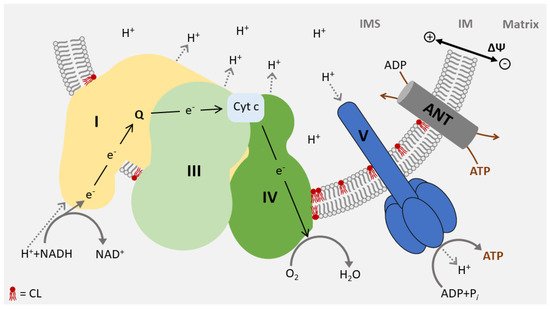
In total, 95% of the cardiac energy demand is covered by oxidative phosphorylation of the respiratory chain in the inner membrane, consisting of five complexes (I–V). Four complexes (I–IV) transport electrons from reducing equivalents (NADH, FADH
2) to oxygen, forming water. The resulting energy is stored in a membrane potential across the inner membrane and converted to ATP by the last complex in the chain (V). The structure of the respiratory chain has been resolved and specific interaction sites for CL were identified in all respiratory chain complexes
[27][41][36,49]. Correspondingly, CL is a structural component of the respiratory chain and is essential for its integrity and full enzymatic activity
[42][43][44][45][50,51,52,53]. Changes in the CL pool, including the accumulation of MLCL, may interfere with the structure of respiratory chain complexes. MLCL, which strongly accumulates in Barth syndrome, binds to complex IV with a substantially reduced affinity and significantly lowers the enzymatic activity of this complex
[46][54]. The structure of the dimeric ATP synthase from bovine mitochondria recently shed light upon the mechanism of proton uptake in the matrix. Interestingly, this study also described the incidence of CL in the integral membrane subunits of complex V
[47][55]. Furthermore, CL is directly involved in the proton export and required for the organization of the F
1F
O ATPase into highly ordered structures
[48][49][56,57].
The respiratory chain complexes assemble into higher-ordered structures—the respirasomes. These supercomplexes include complex I, a dimer of complex III, and one or several complex IV units (
Figure 3). CL molecules are not an only integral to individual complexes but are also associated with the mitochondrial supercomplex formation. It was found to support the structure of supercomplexes and mediate the interaction with the lipid phase of the inner membrane
[50][51][52][58,59,60]. Due to its role in the structure of the respirasomes, CL deficiency causes a defect in respiratory function and a decrease in membrane potential and in ATP synthesis
[27][28][53][36,37,61] (
Table 1).
Figure 3. CL is an essential constituent of respirasomes: The mitochondrial respiratory chain assembles into large oligomeric structures called respirasomes. Respirasomes consist of complex I, a dimer of complex III, and several copies of complex IV. CL is required for respirasome formation and is essential for the activity of the complexes. CL molecules partially interacting with membrane protein complexes are shown in red. ANT, ADP/ATP carrier; Cyt c, Cytochrom c; IM inner membrane; IMS, intermembrane space.
In many forms of cardiac disease, reduced CL levels, alterations in the CL pool or Reactive Oxygen Species (ROS)-induced damage of CL have been observed. Given the important structural function of CL, these changes may have direct structural implications for the respiratory chain. Remodeling of the respiratory chain due to changes in CL has been described in aging, ischaemia/reperfusion and heart failure
[54][55][56][57][62,63,64,65]. These findings are of particular importance as the architecture of respirasomes prevents ROS production. Structural alterations may induce increased ROS generation at the respirasomes, and increased oxidative stress may be an important contributor to the development of heart failure
[58][66]. Therefore, a large number of studies have focused on preventing mitochondrial ROS in various forms of cardiac disease
[59][60][67,68]. These studies, although somewhat inconsistent, showed that reducing ROS levels has the potential to ameliorate ROS-mediated cardiac abnormalities
[61][62][63][69,70,71].