2. Discussion
This study was performed to enable a facile system for monitoring genome editing in living animals as well as to identify edited tissue at a cellular resolution. This work shows that established cre-loxP reporter systems can be used to monitor CRISPR activity. This paper showed that loxP cleavage by SaCas9 can be comparable to cre recombinase activity in deleting stop signals in vitro or in vivo. By using the previously established mT/mG:LSL Luc, it was demonstrated that CRISPR activity could be detected non-invasively in living mice along with a quantitative approach upon dissection of edited tissues.
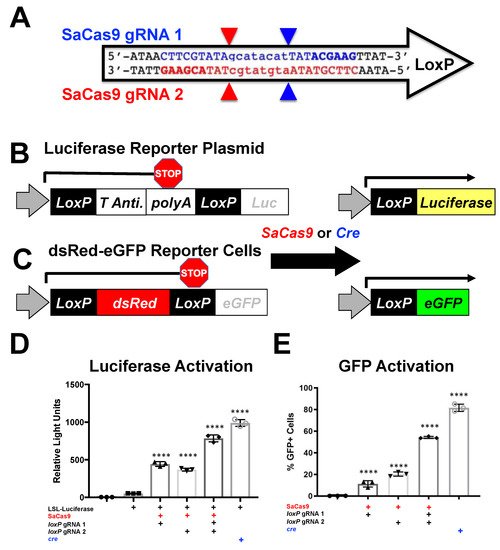
This system also establishes the first
loxP specific reporter for fluorescence monitoring. Previous work has used gRNAs to target adjacent to the
loxP site or within the polyA region (
Supplementary Figure S2A). While these have shown the ability to monitor CRISPR activity via tissue sectioning and DNA analysis, these systems are specific to Ai9 mice and a few related strains and would not be broadly applicable to other reporter systems such as the LSL-mice or the mT/mG target loci
[13]. Other work has shown in vitro targeting with partial overlap with
loxP and complete coverage with the gRNA in the
loxP mutants
lox71 and
lox66 (
Supplementary Figure S2B)
[14]. These previous gRNAs would not be applicable to most mouse models that likely will not have convenient adjacent sequences that allow for targeting and most mouse reporter models do not use
loxP mutants. The system shown herein can be used with any wildtype LoxP site and is therefore capable of being used with any mouse model that uses
loxP to activate or deactivate genes by cre excision. This system could also be used in cases where
loxP is positioned for gene inversion to delete a region or disrupt this process.
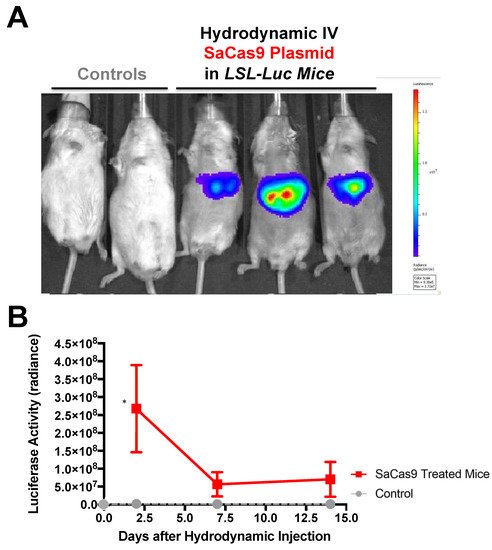
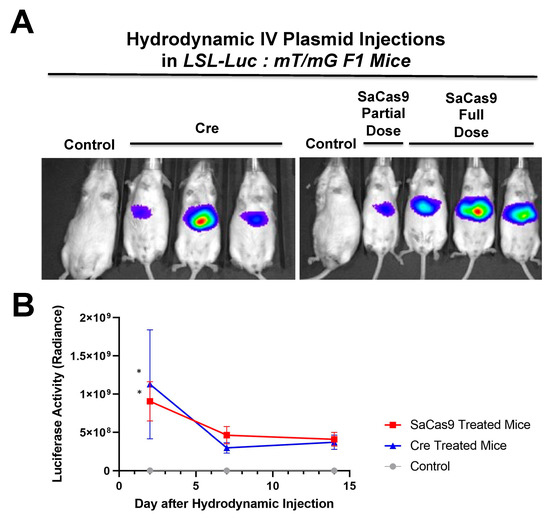
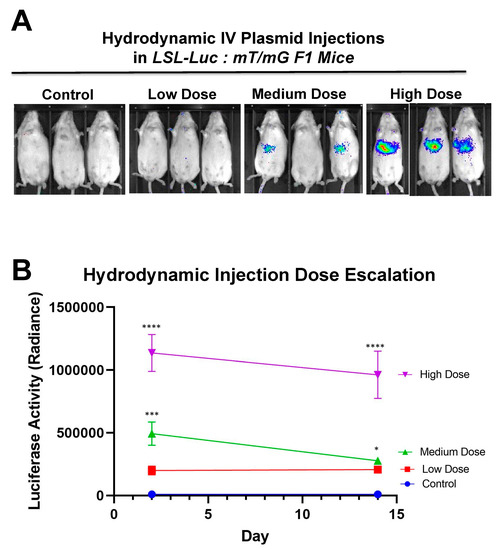
Hydrodynamic delivery functions by rupturing the cell membranes under pressure, pushing the plasmid DNA into the cell. This pressure and resulting damage could be responsible for the decline in luciferase activity from Day 2 to Day 7 as seen in Figure 4, Figure 2 and Figure 5. This coupled with the introduction of neo-antigens in the form of SaCas9 or cre could lead to immunological responses to transduced cells as well. Additionally, there is a discrepancy between the SaCas9 hydrodynamically injected mice in Figure 2; Figure 3. Older mice are not as effective for hydrodynamic delivery and older mice were used in Figure 2. Different maxi-preps were used between the experiments so this may have also contributed.
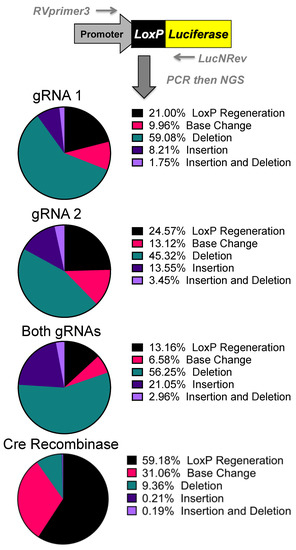
Figure 4. Sequencing of Repair Juncture. DNA was harvested from the previous transfections and amplified using primers in the SV40 promoter and luciferase with next generation sequencing (NGS) sequencing adapters. Using Amplicon-EZ through Genewiz, the deletion results were analyzed and quantified (n = 1).
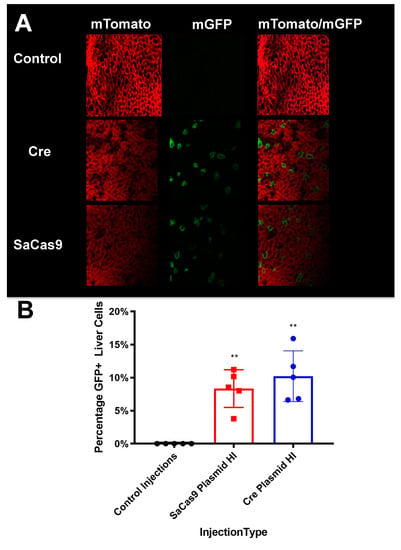
Figure 5. Liver sectioning from hybrid mice. (A) Mouse livers were harvested, fixed, and imaged for tdTomato and GFP activity. Representative microscopy shows cre- and SaCas9-treated mouse livers. (B) Converted cells were counted as a percentage of total cells in representative images across the gene-edited mice. The bars are 95% confidence intervals and were found significant compared to the control by one-way ANOVA and Tukey’s multiple comparisons. The edited groups were not significant compared to each other (n = 5) (** p < 0.005).
Additionally,
Figure 6 shows that there is a dose response to differing amounts of SaCas9. This is of value because it shows that this system could be used to monitor variant levels of delivery via luciferase activity. Whether SaCas9 is delivered by nanoparticle, virus, ribonucleoprotein, or some novel method, this provides a method to detect variations in delivery efficiency non-invasively and with cellular specificity. It should also be noted that the luciferase levels showed a significant decrease compared to previous experiments. We believe this to be the result of imaging differences between the Xenogen and the Lumina.
Supplementary Figure S3 also shows that this reporter system works in tissues beyond the liver. The Barry lab has also previously published in Hillestad et al. that they were able to detect cre recombinase activity using this reporter mouse in the liver, heart, lungs, muscles, brain, kidney, and spleen
[10].
Figure 6. Hydrodynamic dose variation injections. (A) Mice were hydrodynamically injected at varying doses of 7.5 μg, 25 μg, and 83.3 μg of plasmid. Day 2 mouse images are shown. (B) Radiance levels emitting from the mouse livers were measured and compared. Each day was analyzed by one-way ANOVA and Tukey’s multiple comparison. High and medium doses were significant compared to the control on both days while low dose was not significant compared to the control on either day via one-way ANOVA with multiple comparison. Via one-way ANOVA, high, medium, and low doses were significant compared to each other on Day 2. On Day 14 and by one-way ANOVA with Tukey’s multiple comparisons high dose was significant compared to all other groups. Medium dose was only significant compared to the control dose on Day 14. All other comparisons on Day 14 were not significant by one-way ANOVA with Tukey’s multiple comparisons (n = 3) (* p < 0.05, *** p < 0.0005, **** p < 0.0001).
Next generation sequencing analyses of the gene editing outcomes provide some insight into how the cell repairs these DNA breaks prior to reporter activation. Three repair pathways were non-homology-induced repairs, small local homology (microhomology), and homology-based outcomes, especially considering that deletions occur between two identical loxP sequences. Analyses of deletions induced by individual gRNAs demonstrated that most of the edits result in the recreation of a loxP site. Unfortunately, these can be the result of either NHEJ or homology-based repair when using individual gRNAs (Supplementary Figure S1).
Analysis of the dual gRNA-induced deletions demonstrated a different mix of outcomes (Supplementary Figure S1). After the repair pathway-ambiguous loxP recreation, the next two top reads consisted of NHEJ-based events that are caused by different gRNAs targeting each loxP site. Although the top reads are ambiguous, the top reads that are capable of being definitively linked to a pathway are NHEJ. Depending on which gRNA edits which loxP site will determine whether an 8bp region would be duplicated or deleted. That being said, these cuts have the potential to cause microhomology-based events, only one of which is distinguishable as such. The strong prevalence of NHEJ-specific events strongly suggests that this is a major repair mechanism, but with the top detected outcome being ambiguous and repeated cutting of regenerated loxP sites confusing the data further, it cannot be determined at this time what is the primary repair pathway. Further parsing of the mechanism may be done in future work by specifically knocking down proteins related to these pathways.
The sequencing was initially done to explore the possibility of alternative DNA repair pathways being responsible for the increased efficacy of both gRNAs over individual gRNAs. While repair mechanisms cannot be determined conclusively, the increases in efficacy seen with both gRNAs are potentially connected with CRISPR gene editing selecting for mutations that prevent further cutting. With a single gRNA, there is the potential for the gRNA target site to be cut and result in an insertion or deletion (indel) rather than result in a large deletion and preclude the possibility for a subsequent DSB generating a large deletion. An indel near the cut site of the gRNA target site would greatly reduce, if not inhibit, SaCas9 cutting as seen by some of the top reads in NGS
[16]. If a
loxP site is mutated to prevent CRISPR cutting at both
loxP sites, large deletion of the stop cassette would be prevented. With two potential gRNA targets, the potential indel would be further away from the PAM and less likely to inhibit SaCas9 binding
[17]. This may potentially explain the increased efficacy seen with two gRNAs rather than one.
By targeting Cas9 to the loxP site, there is also the opportunity to enable further manipulation of these sites. These models can be used in conjunction with targeted insertion technology to deliver genes of interest at loxP sites. This could be done to modify the sites to express a different gene or to reconstitute stop cassettes with mutant loxP sites that are resistant to CRISPR cutting but available to cre recombinase. There is also the potential of creating a three-outcome cassette: starting cassette, cre recombinase-treated expression cassette, and CRISPR-treated cassette. This would give greater control over animal models that would be able to turn on defective genes via cre recombinase and then deactivate them by CRISPR. There is also the potential of using the deactivated CRISPR enzyme as an inhibitor of cre recombinase, binding loxP and preventing binding and recombination. This could act to prevent cre recombinase activity in specific cell populations.
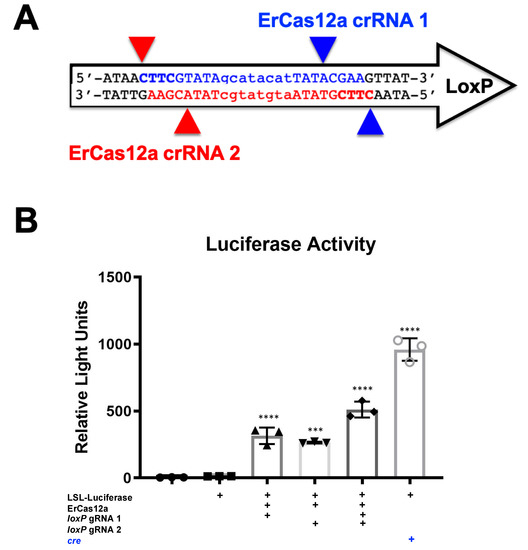
It should be noted that despite the great activity shown by SaCas9 targeting loxP, results may vary depending on the DNA site being targeted or the CRISPR being used. This SaCas9 reporter system is highly efficient while the ErCas12a has lower activity. (Figure 1 and Figure 7). This can be attributed, at least partially, by differential binding and cleavage kinetics demonstrated by Cas12a type effectors compared to Cas9. It would be of interest in the future to determine if similar Cas12a effectors such as AsCas12a or LbCas12a can improve upon this foundational work with SaCas9. It is important to note that this may overestimate the activity when using a gRNA relevant to a gene therapy application or a CRISPR other than SaCas9.
Figure 7. ErCas12a-mediated gene activation through targeting loxP in vitro. (A) The loxP sequence is shown with key features and target sites for CRISPR ErCas12a. The capitalized base pairs are the 13-base-pair palindromic regions flanking the 8-base-pair core that gives loxP its directionality. gRNA 1 homologous strand to the guide RNA is depicted in light blue with the PAM-binding region in dark blue and bold letters and a triangle to indicate the cleavage site. gRNA 2 depicts the same in red. (B) The ErCas12a plasmid co-expressed either gRNA 1 or 2 and was co-transfected with the LSL-Luc reporter plasmid at 2.5 ug. By one-way ANOVA with Tukey’s multiple comparisons, all the groups were significant except between the untransfected control groups (untransfected versus reporter plasmid only) and between the individual gRNA-treated groups (loxP gRNA 1 vs. loxP gRNA 2); 95% confidence intervals are shown (n = 3) (*** p < 0.0005 and **** p < 0.0001).
This three-way reporter system can be applied to in vivo delivery of CRISPR systems to assess the tropism of the delivery system on a broad and narrow level along with a timeline of CRISPR editing. More broadly speaking, this system can be used in combination with any loxP system that relies on deletion for its activity, for which there are over 3000 mice on JAX Laboratories website related to the cre-lox system.
 +1 credit
+1 credit