Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manju Padmasekar Nandigama | + 3797 word(s) | 3797 | 2021-08-10 05:12:56 | | | |
| 2 | Peter Tang | Meta information modification | 3797 | 2021-08-19 02:30:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nandigama, M.P. Exosomes for High-Altitude Epigenetic Research. Encyclopedia. Available online: https://encyclopedia.pub/entry/13303 (accessed on 07 February 2026).
Nandigama MP. Exosomes for High-Altitude Epigenetic Research. Encyclopedia. Available at: https://encyclopedia.pub/entry/13303. Accessed February 07, 2026.
Nandigama, Manju Padmasekar. "Exosomes for High-Altitude Epigenetic Research" Encyclopedia, https://encyclopedia.pub/entry/13303 (accessed February 07, 2026).
Nandigama, M.P. (2021, August 18). Exosomes for High-Altitude Epigenetic Research. In Encyclopedia. https://encyclopedia.pub/entry/13303
Nandigama, Manju Padmasekar. "Exosomes for High-Altitude Epigenetic Research." Encyclopedia. Web. 18 August, 2021.
Copy Citation
Among different difficult environments, high-altitude living is especially demanding because of diminished partial pressure of oxygen and resulting chronic hypobaric hypoxia. This results in poor blood oxygenation and reduces aerobic oxidative respiration in the mitochondria, leading to increased reactive oxygen species generation and activation of hypoxia-inducible gene expression.
high-altitude adaptation
high-altitude pulmonary hypertension
hypobaric hypoxia
epigenetics
exosomes
1. Introduction
Exposomes refers to the totality of environmental exposure individuals experience over their lifetime [1]. Our genome is influenced by exposomes for better or worse. Living organisms at high-altitudes are exposed to an entirely different set of exposomes when compared to lowlanders and this influences their epigenetic make-up. The understanding that organisms are capable of genetically ‘reprogramming’ themselves as well as ‘preprogramming’ future generations to optimize gene expression for a particular environmental signal may have tremendous impact on our perception of both acclimatization and adaptation to hypoxia [2]. High altitude is usually referred to elevation over 2500 m above sea level. Although the percentage of oxygen in air is constant at different altitudes (20.9%), as the vertical height above the earth’s surface increases, the atmospheric pressure (barometric pressure) decreases resulting in decreased partial pressure of inspired oxygen (pO2), leading to less oxygen molecules per breath. For example, at 4000 m altitude, one breath contains just 62% of the oxygen molecules present at sea level. Due to diminished pO2, the driving pressure for gas exchange in the lungs decreases, leading to poor oxygenation of blood as it passes through the pulmonary capillaries. This is more evident during strenuous activity when cardiac output increases. Hence, the blood spends less time at the gas exchanging surface, limiting the time for oxygen diffusion [3]. If an individual from lowland is acutely exposed to high-altitudes (>5500 m), they may lose consciousness, and at over 8000 m, it is an almost certain occurrence. However, when a person is gradually exposed to high-altitudes, they can acclimatize, adapt and survive. During this short-term adaptation phase, the body makes a wide variety of changes to tackle the decreased oxygen in blood [4].
Human survival depends on oxygen homeostasis, which is severely challenged during hypobaric hypoxia. Around 2% of people worldwide, more than 140 million people, live permanently at high altitudes of over 2500 m, such as the Ethiopian Highlands in Africa, the Himalaya Mountains in Asia, and the Andes Mountain Range in South America, where pO2 is low, UV radiation is high, and temperatures are low [5]. Such indigenous populations have developed adaptive features in their morphology, and also in physiological, biochemical, and molecular regulatory pathways to thrive in hypoxic, high-altitude environments. They show better tolerance to hypoxia and exceptional physical performance at altitude [6][7][8]. However, in some, this physiological adaptation fails and hypoxia triggers in them a maladaptive response that leads to various forms of acute and chronic high-altitude illness, such as high-altitude pulmonary edema or chronic mountain sickness [9]. Chronic mountain sickness or Monge’s disease was first described by Carlos Monge in the Andes in 1925 and is the result of failure to physiologically adapt to high-altitude exposure. Chronic mountain sickness occurs in natives or lifelong high-altitude residents, and is characterized by excessive erythrocytosis and severe hypoxemia. Oftentimes, it is associated with moderate or severe pulmonary hypertension (PH) that may evolve to right ventricular hypertrophy and dysfunction, which ultimately leads to congestive heart failure [10][11]. High-altitude pulmonary hypertension (HAPH) is characterized by a mean pulmonary artery pressure >30 mmHg and/or a systolic pulmonary artery pressure >50 mmHg in the absence of excessive erythrocytosis or other lung diseases and might be as frequent as 14% within the population [12]. In cattle, HAPH is known as brisket disease and can be inherited [13][14]. Developmental adaptation to high altitude that happens in natives is different from physiological acclimatization that occurs in sojourners. Native highlanders experience physiological changes to low oxygen for a lifetime, wherein it occurs over days to weeks in the latter. Notably, developmental adaptation is imprinted in the genome and is carried on to future generations, whereas acclimatization to high altitudes is short-lived and is reversible upon return to low altitudes [15].
The lungs play an important role in the acclimatization (short-term adaptation) of the human body to low oxygen at increased altitudes. During acute exposure of lungs to altitude, there is extravascular fluid accumulation in the pulmonary interstitium, and hence, a reduction in lung volume. In this scenario, hyperventilation, together with increased heart rate, helps to supply adequate oxygen to tissues. At rest, increased oxygen demand was met by firstly increasing the tidal volume, at least up to 3500 m [16][17]. Pulmonary circulation responds to hypobaric and normobaric hypoxia by increasing pulmonary arteriolar resistance, which maintains the ventilation–perfusion ratio during localized alveolar hypoxia [18]. Hypoxic pulmonary vasoconstriction occurs in small pulmonary arterioles and veins of a diameter of <900 µm, the veins accounting for ~20% of the total increase in pulmonary vascular resistance caused by hypoxia [19]. The magnitude of hypoxic pulmonary vasoconstriction differs among humans and this difference could be attributed to genetics and adaptive mechanisms. Chronic exposure to hypoxia as a result of high-altitude living can predispose certain individuals to HAPH due to adaptive mechanisms becoming exaggerated, leading to pulmonary vascular remodeling and development of PH, which places an increased pressure load on the right ventricle leading to right heart failure [20][21]. It must be emphasized that causes other than hypoxia may potentially form the basis of and/or contribute to HAPH, such as chronic heart and lung diseases, thrombotic or embolic disease and some gene mutations or even epigenetic mechanisms, which can actually exacerbate the burden on the already hypoxia exposed lungs. Unfortunately, at present, there are no clinically approved drugs for the therapy of HAPH, although the pathological burden is high [9].
Hypoxemia is not only present in high-altitude dwellers but also amongst critically ill patients, such as in PH associated with lung diseases or patients suffering from viral infections such as SARS-COV-2; yet, optimal management strategies remain uncertain. Mechanisms that lead to beneficial adaptation, as opposed to maladaptation, need to be dissected in order to develop individualized treatment strategies. Identifying genomic and epigenomic differences between lowlanders and those adapted for many centuries to the hypoxia of high-altitude may identify beneficial mechanisms of adaptation for subsequent evaluation in the clinical setting. Studying the physiology of members of indigenous populations who are well-adapted to high-altitude may reveal novel target pathways that are amenable to pharmacological manipulation in the critically ill, because trying to mimic the most effective human hypoxia-tolerant phenotype could provide new directions in identifying biomarkers and metabolic pathways which can be further exploited for patient benefit, and this knowledge can be renewed repeatedly as new technologies evolve.
Genomic variation plays an important role in living organisms’ adaptation to varied environments. Genome-wide association studies (GWAS) have identified several genes that underlie high-altitude adaptive phenotypes, many of which are central components of the transcription factor Hypoxia Inducible Factor (HIF), which plays a pivotal role during hypoxia. Candidate genes which are of positive selection among humans in high-altitude environments are NOS2A (nitric oxide synthase 2), ADRA1b (alpha-1B-adrenergic receptor), EDN1 (endothelin 1), PHD3 (HIF-prolyl hydroxylase 3), VEGF (vascular endothelial growth factor), TNC (tenascin C), CDH1 (cadherin 1), EDNRA (endothelin receptor A), PRKAA1 (protein kinase, AMP-activated, alpha 1 catalytic subunit), ELF2 (E74-like factor 2), and PIK3CA (phosphoinositide-3-kinase, catalytic, alpha polypeptide), including the enzyme EGLN1 (Egl-9 Family Hypoxia Inducible Factor 1), which catalyzes the post-transcriptional formation of 4-hydroxyproline. EGLN1 downregulates HIF targets, including EPO (erythropoietin), which is involved in red blood cell production. Other candidate genes are EPAS1 (endothelial PAS domain protein 1), CYP2E1 (a cytochrome P450 enzyme), EDNRA (Endothelin Receptor Type A), ANGPTL4 (angiopoietin-like 4), and CAMK2D (calcium/calmodulin-dependent protein kinase II delta) [22][23][24][25][26].
2. Epigenetics of Hypoxia
Hypoxic stimulus changes DNA methylation and post-translational histone modifications status of gene promoter or enhancer regions, leading to altered interaction of them with transcriptional machinery. On the other hand, an abundant of non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), either transcriptionally silence or degrade targeted messenger RNAs. These epigenetic modifications cause a change in gene expression without a change in DNA sequence, thus forming an integrated and highly complex regulatory network to create a gene expression pattern which forms the basis of hypoxic adaptation [27][28].
When the metabolic demand for oxygen exceeds the supply, hypoxia ensues. Hypoxia can arise under many pathological states, including inflammatory, fibrotic, ischemic and tumorigenic processes, and also due to environmental conditions, such as high-altitude dwelling. Earlier studies on high-altitude adaptations were focused on phenotypes such as hemoglobin concentration or physical work capacity. The subsequent discovery of HIF [29] has been a breakthrough in our understanding of adaption to high-altitudes. HIFs are the master regulator of hypoxic response and was first identified when investigating the molecular mechanisms of the induction of the hematopoietic growth hormone EPO during hypoxia [30]. HIF is a heterodimeric DNA-binding complex composed of two basic helix-loop-helix proteins, the constitutive HIF-1β and one of either hypoxia-inducible α-subunits, HIF-1α or HIF-2α (also known as EPAS1). HIF-β subunits are non-oxygen-responsive nuclear proteins, while HIF-α subunits are highly inducible by hypoxia. Under normoxic conditions, HIF-1α is degraded through hydroxylation, and hence, it is short lived [31]. However, during hypoxia, the α/β heterodimer binds to a core pentanucleotide sequence (RCGTG) in the hypoxia-response elements (HREs) of target genes, resulting in transcriptional regulation [29][32]. In that way, activated HIF-1α directly or indirectly controls the expression of more than 100 genes, and thus, mediates a wide range of physiological and cellular mechanisms necessary to adapt to reduced oxygen [33].
HIF and epigenetic events work together in coordinating hypoxic response pathways and also in the maintenance of the post-hypoxic phenotype. Epigenetics play four key roles during hypoxia: (i) epigenetic modifiers modulate HIF-1α mRNA or protein stability, such as hypermethylation of HIF regulators—von Hippel-Lindau (VHL) and PHD3—having a direct impact on hypoxic signaling, (ii) epigenetic modifiers, via interacting with HIF or modifying HRE binding sites, may also fine-tune HIF-dependent transcriptional programs; for example, HIF-1α associates directly with CREB-Binding-Protein/p300, and thus, augments HIF-1 transcriptional activity and participates in the co-activation of hypoxia inducible genes [34], (iii) the interplay between HIF and epigenome is bi-directional, as hypoxia/HIF leads to a significant global change in histone modifications and DNA methylation in the genome in response to hypoxic exposure, and (iv) the expression of different histone-modifying enzymes are also found to be direct HIF-1 target genes [35].
Notably, when cells were permanently maintained in hypoxia (1% O2), a global change in histone acetylation and DNA methylation was observed. Specifically, H3K9 acetylation and DNA methylation increased in the absence of HIF-1α along with a significant increase in the expression of DNA methyl transferase 3b (DNMT3b) [36]. Hypoxia induces a novel signature of histone modifications, including an increase in histone 3 lysine 4 trimethylation (H3K4me3) levels and a decrease in histone 3 lysine 27 trimethylation (H3K27me3) levels at the promoters of hypoxia-responsive genes and was also found to bind to genes which are marked with histone H3 lysine4 trimethylation (H3K4me3) [37][38]. Apart from global epigenetic changes, locus-specific epigenetic modulations of HIF regulators, such as hypermethylation of VHL and PHD3 promoters, are seen in certain tumor cells, which in turn amplifies HIF signaling [39][40]. Chronic hypoxia was shown to significantly induce methylation at the CpG site in the VHL promoter, decreased VHL expression, and increased EPAS1 and EPO expression, leading to excessive erythrocytosis in a rat model of chronic mountain sickness. Furthermore, the DNMT inhibitor 5-azacytidine reduced chronic hypoxia-induced erythroid proliferation in the bone marrow of rats with chronic mountain sickness by suppressing VHL methylation and DNMTs expression [41]. Chronic hypoxia was also found to upregulate DNMTs in ovine uterine arteries and thereby repressed large conductance Ca2+-activated K+ channel function [41].
On the other hand, VHL was shown to prevent HIF activation through the recruitment of histone deacetylase enzymes (HDACs) [41]. HDACs 1, 3, and 7 directly bind to HIF-1α, and thus, regulates its function during hypoxic exposure [42]. Sirtuin 1 (SIRT1) is a multifaceted NAD+-dependent protein deacetylase whose expression is increased during hypoxia [43]. SIRT1 modulates cellular responses to hypoxia by deacetylating HIF-1α and EPAS1, which leads to totally different outcomes: repression of HIF-1α activity and activation of EPAS1 signaling, respectively [44][45]. SIRT1-mediated deacetylation also suppresses the functions of tumor suppressor p53, which is stabilized and activated during hypoxic conditions. The p53KO mice was prone to pulmonary vascular remodeling and increased pulmonary arterial smooth muscle cell proliferation when exposed to chronic hypoxia leading to PH [46]. Hypoxia induced upregulation of HIF-1α is also found to activate Jumonji proteins (JMJDs), which possess histone demethylase properties [47]. It was also shown that high density methylation of the 5′-untransplated region (5′-UTR) of EPO controls its tissue specific expression and hypoxia activates Histone H3K9 demethylase JMJD1A leading to EPO expression [48][49]. Several histone lysine demethylases (KDMs) are modulated under hypoxia upon HIF-1α stabilization.
MicroRNAs are small RNA molecules, ∼22 nucleotides long, that can negatively control their target gene expression post-transcriptionally [50]. Recent evidence suggests that miRNAs are key elements in the response to hypoxia, regulating gene expression through post-transcriptional mechanisms. MicroRNAs are also important modulators of DNMT3a and DNMT3b expression under hypoxic conditions. A decreased expression of miR-29 correlates with increased DNMT3 expression in lung cancer cells [51], and it was shown that miR-29 levels fall significantly in ischemic hearts [52]. This might indicate that miR-29 controls DNMT3 expression under hypoxic conditions. miR-210 (hypoximir) is a well-known miRNA induced under hypoxic condition in several types of tissues and cells, and its targets control mitochondrial metabolism, angiogenesis, DNA damage response, apoptosis, and cell survival, and contributes to cellular adaptation to hypoxia [53][54]. These observations suggest the involvement of epigenetic processes in HIF-1-associated gene signaling in response to hypoxia, and may also control other unknown genomic processes. Thus, we can safely presume that exposure to high altitude may also trigger similar epigenetic mechanisms, leading to the adaptation of living beings to continuous hypobaric hypoxia. Accordingly, whole-genome DNA methylation data generated from heart tissues of Tibetan pigs grown in the highland and lowland, as well as Yorkshire pigs grown in the highland and lowland, using methylated DNA immunoprecipitation sequencing, showed several thousand differentially methylated regions (DMRs) containing many differentially methylated genes. Enrichment of DMRs associated with HIF-1 signaling pathway and pathways involved in hypoxia-related processes has been observed. Hence, they were considered to be important candidate genes for high-altitude adaptation in Tibetan pigs [55]. Apart from miRNAs, lncRNAs role in hypoxic regulation is increasingly evident in the field of cancer biology. lncRNAs do not code for proteins and are more than 200 nucleotides in length; they act in many different ways to modify gene expression. In cancer, they are found to act as enhancers of HIF-1α and EPAS1 expression and as well regulators of HIF stability and transcriptional activity [56].
3. Epigenetic Adaptation of Animals to High Altitude
Although the mechanism of high-altitude adaptation which may involve complex multi-gene expression and interaction is not fully understood, modern multi-omics approaches allow for the identification of key functional genes, and their regulatory mechanisms including epigenetic modifications underlying high-altitude adaptation (Figure 1). High-altitude adaptation is a classic example of selective pressure shaping the human genome [57]. Genetic variations alone cannot explain all the biological variations [58] that leads to adaptive or maladaptive processes that happen in high-altitude dwellers. DNA methylation patterns established prenatally as well as early in the postnatal period are associated with methylation patterns later in life, and thus, they may be involved in the organism’s phenotypic response to their environment. This phenomenon is known as the predictive-adaptive response, which helps the cells to be ‘prepared’ long after the stimulus which caused the epigenetic mark is gone, thus providing cells with a memory about its previous experiences [59]. This provides the cells with an ability to adapt to a new environment when genetic variation is limited. Hence, epigenetic changes can potentially act as mechanisms of phenotypic plasticity [60] through altered biological pathways that improve reproductive fitness and survival under conditions of chronic hypoxia, and studying them may provide a means of identifying candidate biochemical markers of successful adaptation to high altitude, and thereby, lead to viable pharmacological interventions that may benefit hypoxemic patients.
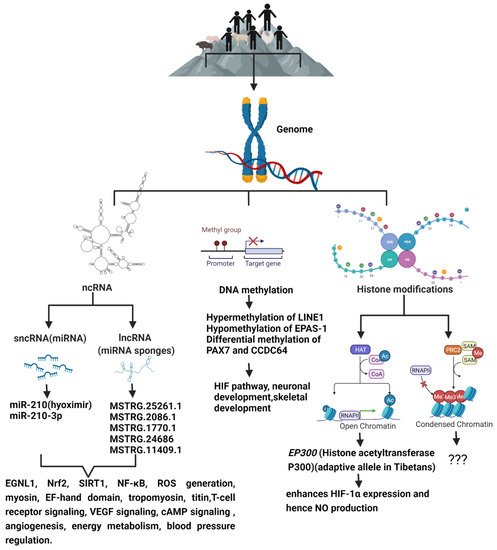
Figure 1. The schematic diagram shows the epigenetic modifications predominant in high-altitude living organisms when compared to their lowland counterparts, and the genes and signaling pathways that are altered as a result of high-altitude living.
4. Epigenetics of High-Altitude-Induced (Hypobaric Hypoxia-Induced) Pulmonary Hypertension
PH is a complex, chronic, progressive, severely debilitating, incurable, and life-threatening disease of the vascular and non-vascular components of lung leading to hemodynamic alteration of pulmonary circulation. As a result, PH patients suffer from increased pulmonary arterial pressure due to increased vascular resistance, which eventually culminates in right ventricular dysfunction and failure due to pressure overload. PH occurs due to a variety of genetic and pathogenic causes and also due to the environmental conditions to which one is exposed [61] and is characterized by pulmonary vasoconstriction and abnormal (“pseudo-malignant”) inward remodeling processes that may affect all vessel layers (intima, media, and adventitia) [62][63].
Global DNA methylation was increased in PH rat models after hypobaric hypoxia exposure. DNMT3B was upregulated in both PH patients and rodent models. Furthermore, Dnmt3b−/− rats exhibited more severe pulmonary vascular remodeling and inhibition of DNMT3B promoted proliferation/migration of PASMCs in response to PDGF-BB. In contrast, overexpressing DNMT3B in PASMCs was found to attenuate PDGF-BB-induced proliferation/migration and ameliorated hypoxia-mediated PH and right ventricular hypertrophy in mice. DNMT3B was also shown to transcriptionally regulate inflammatory pathways [64].
PH is also a hallmark of High-Altitude Pulmonary Edema (HAPE). Aberrant methylation in the apelin system could predict the risk of HAPE. HIF stimulates the secretion of apelin, a potent vasodilator during hypobaric hypoxia. HAPE patients show decreased apelin and nitrite levels as opposed to humans who are well-adapted to hypoxia. The increased methylation of the apelin CpG island, and hence, a lower secretion of apelin and, consequently, lower NO levels, could contribute to the pathology of HAPE due to impaired vasodilation [65].
The above-mentioned studies point to the probable role of epigenetic alterations in the genome of high-altitude living beings and in the pathology of HAPH. Contrasting to genetic mutations, epigenetic changes are pharmacologically reversible, making them an attractive target as therapeutic strategies for HAPH.
5. Exosomes and Hypoxia
Exosomes are the smallest type of extracellular vesicles (30–100 nm) released by almost all cells of the human body both in health as well as in diseased conditions [66]. During its initial discovery, they were thought to carry unwanted waste materials. However, later studies show that they act as messengers communicating between cells of different tissues by influencing the physiological and metabolic pathways of recipient cells [67]. Exosomes carry varied and useful cargo load, which includes cell membrane proteins, enzymes, growth factors, cytokines, DNA, mitochondrial DNA (mtDNA), mRNAs, lncRNAs, miRNAs, small nucleolar RNAs (snoRNAs), and lipids. They also carry signaling molecules and help in tissue remodeling [68]. The field of exosomes is a rapidly growing area in basic research, biomarker discovery, as well as in regenerative therapy, and even in nano biotechnology, as nanocarriers of drugs because of their viral-like transfection efficiency and inherent biological functions. Exosomes are enriched with molecules such as CD63, CD9, and CD81, Alix, and TSG101, which serve as markers for exosomes; however, the distribution of these surface molecules varies greatly depending on the cell type [66]. The exosomes are also a source of several potential biomarkers that are well correlated with the disease’s severity [69].
The lungs are a complex organ with a wide variety of cell types. Hence, exosomes are thought to play an important role in lungs during cell–cell communication. The lungs are also the organ with the highest vascular density in human body. It is very much understandable that the lungs contribute to the majority of exosomes that are circulating in the blood. Exosomes are released by a wide range of cell types present within the lungs, including endothelial cells, stem cells, epithelial cells, alveolar macrophage, and tumor cells [70]. During disease conditions, this exosomal load can be higher or lower depending on the stimuli, and the exosomal cargo may also vary widely depending on the infecting organism, immune system players, and the inflammatory microenvironment [71].
6. Exosome for High-Altitude Epigenetic Research
Exosomes are secreted by most cells of humans and are found abundantly in body fluids, such as saliva, plasma, serum, cerebrospinal fluid, and urine [72][73][74][75][76]. Hence, exosomes can be isolated easily from plethora of body fluids—thus, they are easily sampleable. In cancer biology, liquid biopsies have revolutionized the field of biomarker discovery, and exosomes play a pivotal role [77][78][79]. Exosomes from liquid biopsies are extremely stable and their phospholipid bilayer protects their bioactive cargo from degradation [80][81]. DNA methylation appears to be cell-type specific, which poses challenge in human studies done using whole blood because sampling is usually limited to the analysis of peripheral blood cells, which may or may not reflect systemic adaptations. Exosomes can overcome this challenge as their isolation and analytic techniques has gotten more and more refined. Exosomes from serum or plasma also reflect the exosomal content from many different tissues and cells of body. However, advancement in exosomal biology allows us to identify and separate exosomes from a particular tissue by its membrane protein content [82], and hence, studying their genomic content can be more specific and fruitful in terms of understanding a specific organ’s epigenetic profile during health and disease rather than from whole blood.
Exosomes carry unique sets of mRNAs, rRNAs, miRNAs, lncRNAs, and other small non-coding RNAs (ncRNAs, e.g., piRNAs, snRNAs, snoRNAs, scaRNAs, and Y-RNAs) [83][84]. Some miRNAs that are abundant in parent cells are also most commonly found in exosomes, hence making them excellent tools for biomarker discovery. However, this is not always the case [83][85]. The majority of microRNAs detectable in serum and saliva were found to be concentrated in exosomes [86]. It has been reported that the absolute amounts of miRNAs are estimated to range 1 × 102–1 × 105 copies per cell [87], while a single exosome can carry up to approximately 500 copies of miRNAs [87]. With the current advances in next-generation deep sequencing (NGS), the entire spectrum of known and novel miRNA can be profiled with minimal RNA input and hence analysis of exosomal RNA has become much feasible [88]. The most important limitation in working with exosomes is their nanosize, and hence, the laborious and time-consuming ultracentrifugation steps that are involved in their isolation. However, recent advancements in the exosomal isolation kits that are available in market has decreased the burden of extensive ultracentrifugal steps without having to compromise exosomal quality [89]. The coming years might prove to be comparatively trouble-free and cost-effective as far as exosome isolation is concerned. International Society for Extracellular Vesicles provides guidelines to be followed for the publication of EV (extracellular vesicle) studies [90].
All this makes exosomes a good tool for epigenetic research, and it would be very helpful to discover epigenetic adaptive and maladaptive pathways in indigenous populations occupying high-altitude environments or in sojourners (Figure 2).
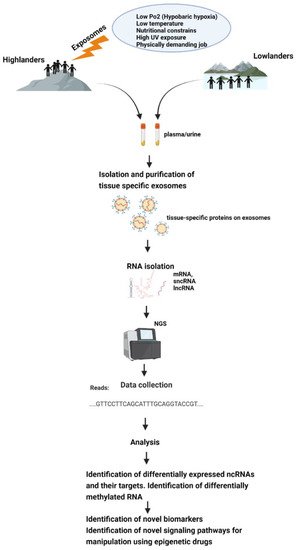
Figure 2. Schematic representation of how exposomes affect the epigenome of high-altitude dwelling living organisms—which could be studied using exosomes.
References
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850.
- Brown, C.J.; Rupert, J.L. Hypoxia and environmental epigenetics. High Alt. Med. Biol. 2014, 15, 323–330.
- Petersson, J.; Glenny, R.W. Gas exchange and ventilation-perfusion relationships in the lung. Eur. Respir. J. 2014, 44, 1023–1041.
- Imray, C.; Wright, A.; Subudhi, A.; Roach, R. Acute mountain sickness: Pathophysiology, prevention, and treatment. Prog. Cardiovasc. Dis. 2010, 52, 467–484.
- Beall, C.M. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr. Comp. Biol. 2006, 46, 18–24.
- Petousi, N.; Robbins, P.A. Human adaptation to the hypoxia of high altitude: The Tibetan paradigm from the pregenomic to the postgenomic era. J. Appl. Physiol. (1985) 2014, 116, 875–884.
- Cheviron, Z.A.; Brumfield, R.T. Genomic insights into adaptation to high-altitude environments. Heredity 2012, 108, 354–361.
- Frisancho, A.R. Developmental functional adaptation to high altitude: Review. Am. J. Hum. Biol. 2013, 25, 151–168.
- Sydykov, A.; Mamazhakypov, A.; Maripov, A.; Kosanovic, D.; Weissmann, N.; Ghofrani, H.A.; Sarybaev, A.S.; Schermuly, R.T. Pulmonary Hypertension in Acute and Chronic High Altitude Maladaptation Disorders. Int. J. Environ. Res. Public Health 2021, 18, 1692.
- Arias-Stella, J.; Kruger, H.; Recavarren, S. Pathology of chronic mountain sickness. Thorax 1973, 28, 701–708.
- Garrido, E.; Botella de Maglia, J.; Castillo, O. Acute, subacute and chronic mountain sickness. Rev. Clin. Esp. 2020.
- Brito, J.; Siques, P.; Pena, E. Long-term chronic intermittent hypoxia: A particular form of chronic high-altitude pulmonary hypertension. Pulm. Circ. 2020, 10, 5–12.
- Newman, J.H.; Holt, T.N.; Hedges, L.K.; Womack, B.; Memon, S.S.; Willers, E.D.; Wheeler, L.; Phillips, J.A., 3rd; Hamid, R. High-altitude pulmonary hypertension in cattle (brisket disease): Candidate genes and gene expression profiling of peripheral blood mononuclear cells. Pulm. Circ. 2011, 1, 462–469.
- Shirley, K.L.; Beckman, D.W.; Garrick, D.J. Inheritance of pulmonary arterial pressure in Angus cattle and its correlation with growth. J. Anim. Sci. 2008, 86, 815–819.
- Jungmann, H. Acclimatization and adaptation to high altitude. Prog. Biometeorol. 1974, 1 Pt 1B, 474–481, 695–698.
- Cogo, A. The lung at high altitude. Multidiscip. Respir. Med. 2011, 6, 14–15.
- Cremona, G.; Asnaghi, R.; Baderna, P.; Brunetto, A.; Brutsaert, T.; Cavallaro, C.; Clark, T.M.; Cogo, A.; Donis, R.; Lanfranchi, P.; et al. Pulmonary extravascular fluid accumulation in recreational climbers: A prospective study. Lancet 2002, 359, 303–309.
- Aaronson, P.I.; Robertson, T.P.; Knock, G.A.; Becker, S.; Lewis, T.H.; Snetkov, V.; Ward, J.P. Hypoxic pulmonary vasoconstriction: Mechanisms and controversies. J. Physiol. 2006, 570, 53–58.
- Hakim, T.S.; Michel, R.P.; Minami, H.; Chang, H.K. Site of pulmonary hypoxic vasoconstriction studied with arterial and venous occlusion. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 54, 1298–1302.
- Stream, J.O.; Luks, A.M.; Grissom, C.K. Lung disease at high altitude. Expert Rev. Respir. Med. 2009, 3, 635–650.
- Naeije, R. Pulmonary circulation at high altitude. Respiration 1997, 64, 429–434.
- Bigham, A.W.; Mao, X.; Mei, R.; Brutsaert, T.; Wilson, M.J.; Julian, C.G.; Parra, E.J.; Akey, J.M.; Moore, L.G.; Shriver, M.D. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genom. 2009, 4, 79–90.
- Simonson, T.S.; Yang, Y.; Huff, C.D.; Yun, H.; Qin, G.; Witherspoon, D.J.; Bai, Z.; Lorenzo, F.R.; Xing, J.; Jorde, L.B.; et al. Genetic evidence for high-altitude adaptation in Tibet. Science 2010, 329, 72–75.
- Beall, C.M.; Cavalleri, G.L.; Deng, L.; Elston, R.C.; Gao, Y.; Knight, J.; Li, C.; Li, J.C.; Liang, Y.; McCormack, M.; et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA 2010, 107, 11459–11464.
- Bigham, A.; Bauchet, M.; Pinto, D.; Mao, X.; Akey, J.M.; Mei, R.; Scherer, S.W.; Julian, C.G.; Wilson, M.J.; Lopez Herraez, D.; et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010, 6, e1001116.
- Yang, J.; Jin, Z.B.; Chen, J.; Huang, X.F.; Li, X.M.; Liang, Y.B.; Mao, J.Y.; Chen, X.; Zheng, Z.; Bakshi, A.; et al. Genetic signatures of high-altitude adaptation in Tibetans. Proc. Natl. Acad. Sci. USA 2017, 114, 4189–4194.
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquiere, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68.
- Xin, J.; Zhang, H.; He, Y.; Duren, Z.; Bai, C.; Chen, L.; Luo, X.; Yan, D.S.; Zhang, C.; Zhu, X.; et al. Chromatin accessibility landscape and regulatory network of high-altitude hypoxia adaptation. Nat. Commun. 2020, 11, 4928.
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237.
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell Biol. 1992, 12, 5447–5454.
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182.
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12.
- Weidemann, A.; Johnson, R.S. Biology of HIF-1alpha. Cell Death Differ. 2008, 15, 621–627.
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15.
- Watson, J.A.; Watson, C.J.; McCann, A.; Baugh, J. Epigenetics, the epicenter of the hypoxic response. Epigenetics 2010, 5, 293–296.
- Watson, J.A.; Watson, C.J.; McCrohan, A.M.; Woodfine, K.; Tosetto, M.; McDaid, J.; Gallagher, E.; Betts, D.; Baugh, J.; O’Sullivan, J.; et al. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum. Mol. Genet. 2009, 18, 3594–3604.
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 2008, 640, 174–179.
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298.
- Place, T.L.; Fitzgerald, M.P.; Venkataraman, S.; Vorrink, S.U.; Case, A.J.; Teoh, M.L.; Domann, F.E. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS ONE 2011, 6, e14617.
- Yang, L.; Zhao, Z.; Zhao, S.; Chen, C.; Cong, X.; Li, Z.; Ren, M. The Clinicopathological Significance of Epigenetic Silencing of VHL Promoter and Renal Cell Carcinoma: A Meta-Analysis. Cell Physiol. Biochem. 2016, 40, 1465–1472.
- Yang, M.; Zhu, M.; Song, K.; Wuren, T.; Yan, J.; Ge, R.L.; Ji, L.; Cui, S. VHL gene methylation contributes to excessive erythrocytosis in chronic mountain sickness rat model by upregulating the HIF-2alpha/EPO pathway. Life Sci. 2021, 266, 118873.
- Liang, D.; Kong, X.; Sang, N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle 2006, 5, 2430–2435.
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011, 286, 13869–13878.
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878.
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293.
- Mizuno, S.; Bogaard, H.J.; Kraskauskas, D.; Alhussaini, A.; Gomez-Arroyo, J.; Voelkel, N.F.; Ishizaki, T. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L753–L761.
- Lee, H.Y.; Choi, K.; Oh, H.; Park, Y.K.; Park, H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol. Cells 2014, 37, 43–50.
- Tian, Z.; Yao, L.; Shen, Y.; Guo, X.; Duan, X. Histone H3K9 demethylase JMJD1A is a co-activator of erythropoietin expression under hypoxia. Int. J. Biochem. Cell Biol. 2019, 109, 33–39.
- Steinmann, K.; Richter, A.M.; Dammann, R.H. Epigenetic silencing of erythropoietin in human cancers. Genes Cancer 2011, 2, 65–73.
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24R–29R.
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810.
- Ye, Y.; Hu, Z.; Lin, Y.; Zhang, C.; Perez-Polo, J.R. Downregulation of microRNA-29 by antisense inhibitors and a PPAR-gamma agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2010, 87, 535–544.
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210–micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237.
- Huang, X.; Zuo, J. Emerging roles of miR-210 and other non-coding RNAs in the hypoxic response. Acta Biochim. Biophys. Sin. 2014, 46, 220–232.
- Zhang, B.; Ban, D.; Gou, X.; Zhang, Y.; Yang, L.; Chamba, Y.; Zhang, H. Genome-wide DNA methylation profiles in Tibetan and Yorkshire pigs under high-altitude hypoxia. J. Anim. Sci. Biotechnol. 2019, 10, 25.
- Barth, D.A.; Prinz, F.; Teppan, J.; Jonas, K.; Klec, C.; Pichler, M. Long-Noncoding RNA (lncRNA) in the Regulation of Hypoxia-Inducible Factor (HIF) in Cancer. Noncoding RNA 2020, 6, 27.
- Hancock, A.M.; Alkorta-Aranburu, G.; Witonsky, D.B.; Di Rienzo, A. Adaptations to new environments in humans: The role of subtle allele frequency shifts. Philos. Trans. R. Soc. Lond. B Biol Sci. 2010, 365, 2459–2468.
- Audergon, P.N.; Catania, S.; Kagansky, A.; Tong, P.; Shukla, M.; Pidoux, A.L.; Allshire, R.C. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 2015, 348, 132–135.
- Gluckman, P.D.; Hanson, M.A.; Spencer, H.G. Predictive adaptive responses and human evolution. Trends Ecol. Evol. 2005, 20, 527–533.
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373.
- Shimoda, L.A.; Laurie, S.S. Vascular remodeling in pulmonary hypertension. J. Mol. Med. 2013, 91, 297–309.
- Merklinger, S.L.; Jones, P.L.; Martinez, E.C.; Rabinovitch, M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation 2005, 112, 423–431.
- Sakao, S.; Tatsumi, K.; Voelkel, N.F. Endothelial cells and pulmonary arterial hypertension: Apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 2009, 10, 95.
- Yan, Y.; He, Y.Y.; Jiang, X.; Wang, Y.; Chen, J.W.; Zhao, J.H.; Ye, J.; Lian, T.Y.; Zhang, X.; Zhang, R.J.; et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci. Adv. 2020, 6, eaba2470.
- Mishra, A.; Kohli, S.; Dua, S.; Thinlas, T.; Mohammad, G.; Pasha, M.A. Genetic differences and aberrant methylation in the apelin system predict the risk of high-altitude pulmonary edema. Proc. Natl. Acad. Sci. USA 2015, 112, 6134–6139.
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579.
- Bang, C.; Thum, T. Exosomes: New players in cell-cell communication. Int. J. Biochem. Cell Biol. 2012, 44, 2060–2064.
- Das, A.; Mohan, V.; Krishnaswamy, V.R.; Solomonov, I.; Sagi, I. Exosomes as a storehouse of tissue remodeling proteases and mediators of cancer progression. Cancer Metastasis Rev. 2019, 38, 455–468.
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 632290.
- Fujita, Y.; Kosaka, N.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular vesicles in lung microenvironment and pathogenesis. Trends Mol. Med. 2015, 21, 533–542.
- Li, S.P.; Lin, Z.X.; Jiang, X.Y.; Yu, X.Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol. Sin. 2018, 39, 542–551.
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373.
- Hornick, N.I.; Huan, J.; Doron, B.; Goloviznina, N.A.; Lapidus, J.; Chang, B.H.; Kurre, P. Serum Exosome MicroRNA as a Minimally-Invasive Early Biomarker of AML. Sci. Rep. 2015, 5, 11295.
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727.
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887.
- Zlotogorski-Hurvitz, A.; Dayan, D.; Chaushu, G.; Korvala, J.; Salo, T.; Sormunen, R.; Vered, M. Human saliva-derived exosomes: Comparing methods of isolation. J. Histochem. Cytochem. 2015, 63, 181–189.
- Rabinowits, G.; Gercel-Taylor, C.; Day, J.M.; Taylor, D.D.; Kloecker, G.H. Exosomal microRNA: A diagnostic marker for lung cancer. Clin. Lung Cancer 2009, 10, 42–46.
- Wang, J.; Liu, Y.; Sun, W.; Zhang, Q.; Gu, T.; Li, G. Plasma exosomes as novel biomarker for the early diagnosis of gastric cancer. Cancer Biomark. 2018, 21, 805–812.
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182.
- Kalra, H.; Adda, C.G.; Liem, M.; Ang, C.S.; Mechler, A.; Simpson, R.J.; Hulett, M.D.; Mathivanan, S. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 2013, 13, 3354–3364.
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606.
- Wu, D.; Yan, J.; Shen, X.; Sun, Y.; Thulin, M.; Cai, Y.; Wik, L.; Shen, Q.; Oelrich, J.; Qian, X.; et al. Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nat. Commun. 2019, 10, 3854.
- van Balkom, B.W.; Eisele, A.S.; Pegtel, D.M.; Bervoets, S.; Verhaar, M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J. Extracell. Vesicles 2015, 4, 26760.
- Li, M.; Zeringer, E.; Barta, T.; Schageman, J.; Cheng, A.; Vlassov, A.V. Analysis of the RNA content of the exosomes derived from blood serum and urine and its potential as biomarkers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130502.
- Vautrot, V.; Chanteloup, G.; Elmallah, M.; Cordonnier, M.; Aubin, F.; Garrido, C.; Gobbo, J. Exosomal miRNA: Small Molecules, Big Impact in Colorectal Cancer. J. Oncol. 2019, 2019, 8585276.
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, e30679.
- Dilsiz, N. Role of exosomes and exosomal microRNAs in cancer. Future Sci. OA 2020, 6, FSO465.
- Cheng, L.; Quek, C.Y.; Sun, X.; Bellingham, S.A.; Hill, A.F. The detection of microRNA associated with Alzheimer’s disease in biological fluids using next-generation sequencing technologies. Front. Genet. 2013, 4, 150.
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335.
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
More
Information
Subjects:
Agriculture, Dairy & Animal Science
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
19 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No