 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luis D'Marco | + 3292 word(s) | 3292 | 2021-07-15 04:33:30 | | | |
| 2 | Bruce Ren | -21 word(s) | 3271 | 2021-08-10 11:26:26 | | |
Video Upload Options
Diabetes mellitus (DM) is considered one of the most massive epidemics of the twenty-first century due to its high mortality rates caused mainly due to its complications; therefore, the early identification of such complications becomes a race against time to establish a prompt diagnosis. The research of complications of DM over the years has allowed the development of numerous alternatives for diagnosis. Among these emerge the quantification of advanced glycation end products (AGEs) given their increased levels due to chronic hyperglycemia, while also being related to the induction of different stress-associated cellular responses and proinflammatory mechanisms involved in the progression of chronic complications of DM. Additionally, the investigation for more valuable and safe techniques has led to developing a newer, noninvasive, and effective tool, termed skin fluorescence (SAF).
1. Introduction
2. Protein Glycation and Formation of Advanced Glycation End Products
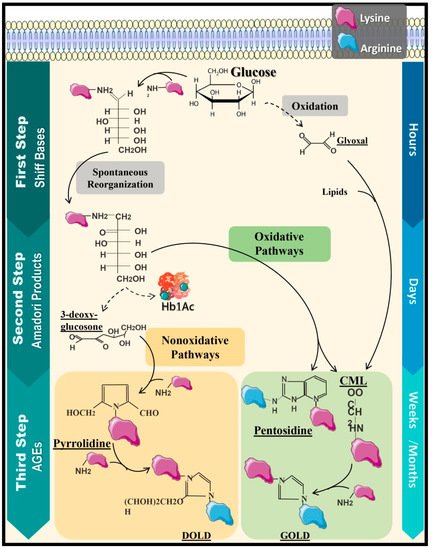
3. AGEs and Their Implication in Chronic Complications of Diabetes Mellitus
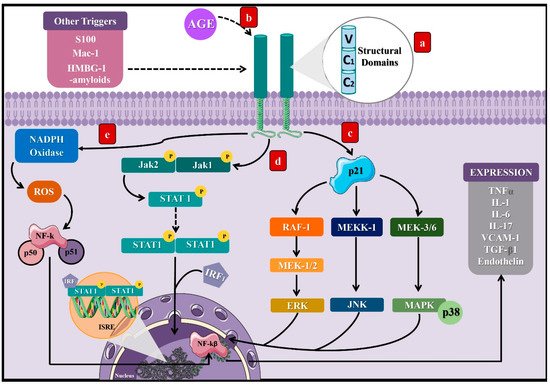
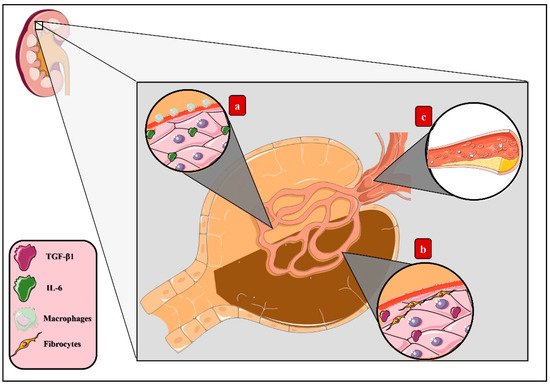
3.1. Molecular Mechanisms of AGEs in Microvascular Complications of DM
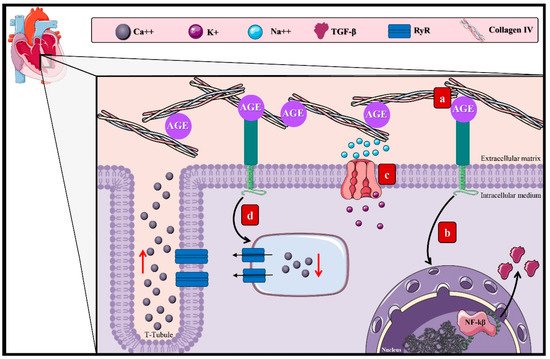
3.2. AGEs and the Macrovascular Alterations in DM
References
- Kerner, W.; Brückel, J. German Diabetes Association Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2014, 122, 384–386.
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes Mellitus Statistics on Prevalence and Mortality: Facts and Fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622.
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015; Available online: https://www.desang.net/2017/11/idf-diabetes-atlas-7th-edition/ (accessed on 7 May 2021).
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharm. 2014, 18, 1–14.
- Butalia, S.; Patel, A.B.; Johnson, J.A.; Ghali, W.A.; Rabi, D.M. Geographic Clustering of Acute Complications and Sociodemographic Factors in Adults with Type 1 Diabetes. Can. J. Diabetes 2017, 41, 132–137.
- Elgart, J.F.; Caporale, J.E.; Asteazarán, S.; De La Fuente, J.L.; Camilluci, C.; Brown, J.B.; González, C.D.; Gagliardino, J.J. Association between Socioeconomic Status, Type 2 Diabetes and Its Chronic Complications in Argentina. Diabetes Res. Clin. Pract. 2014, 104, 241–247.
- Shi, Y.; Vanhoutte, P.M. Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 2017, 9, 434–449.
- Loomis, S.J.; Chen, Y.; Sacks, D.B.; Christenson, E.S.; Christenson, R.H.; Rebholz, C.M.; Selvin, E. Cross-Sectional Analysis of AGE-CML, SRAGE, and EsRAGE with Diabetes and Cardiometabolic Risk Factors in a Community-Based Cohort. Clin. Chem. 2017, 63, 980–989.
- Vélayoudom-Céphise, F.-L.; Rajaobelina, K.; Helmer, C.; Nov, S.; Pupier, E.; Blanco, L.; Hugo, M.; Farges, B.; Astrugue, C.; Gin, H.; et al. Skin Autofluorescence Predicts Cardio-Renal Outcome in Type 1 Diabetes: A Longitudinal Study. Cardiovasc. Diabetol. 2016, 15, 127.
- Thomas, M.C.; Woodward, M.; Neal, B.; Li, Q.; Pickering, R.; Marre, M.; Williams, B.; Perkovic, V.; Cooper, M.E.; Zoungas, S.; et al. Relationship between Levels of Advanced Glycation End Products and Their Soluble Receptor and Adverse Outcomes in Adults with Type 2 Diabetes. Diabetes Care 2015, 38, 1891–1897.
- D’Alessandro, A.; Mirasole, C.; Zolla, L. Haemoglobin Glycation (Hb1Ac) Increases during Red Blood Cell Storage: A MALDI-TOF Mass-Spectrometry-Based Investigation. Vox Sang. 2013, 105, 177–180.
- Cho, Y.H.; Craig, M.E.; Januszewski, A.S.; Benitez-Aguirre, P.; Hing, S.; Jenkins, A.J.; Donaghue, K.C. Higher Skin Autofluorescence in Young People with Type 1 Diabetes and Microvascular Complications. Diabet. Med. 2017, 34, 543–550.
- Botros, N.; Sluik, D.; van Waateringe, R.P.; de Vries, J.H.M.; Geelen, A.; Feskens, E.J.M. Advanced Glycation End-Products (AGEs) and Associations with Cardio-Metabolic, Lifestyle, and Dietary Factors in a General Population: The NQplus Study. Diabetes Metab. Res. Rev. 2017, 33.
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916.
- Mondaca-Navarro, B.A.; Ávila-Villa, L.A.; González-Córdova, A.F.; López-Cervantes, J.; Sánchez-Machado, D.I.; Campas-Baypoli, O.N.; Rodríguez-Ramírez, R. Antioxidant and Chelating Capacity of Maillard Reaction Products in Amino Acid-Sugar Model Systems: Applications for Food Processing. J. Sci. Food Agric. 2017, 97, 3522–3529.
- Brownlee, M.; Vlassara, H.; Cerami, A. Nonenzymatic Glycosylation and the Pathogenesis of Diabetic Complications. Ann. Intern. Med. 1984, 101, 527–537.
- Chu, F.L.; Yaylayan, V.A. Post-Schiff Base Chemistry of the Maillard Reaction: Mechanism of Imine Isomerization. Ann. N. Y. Acad. Sci. 2008, 1126, 30–37.
- Johnson, K.L.; Williams, J.G.; Maleki, S.J.; Hurlburt, B.K.; London, R.E.; Mueller, G.A. Enhanced Approaches for Identifying Amadori Products: Application to Peanut Allergens. J. Agric. Food Chem. 2016, 64, 1406–1413.
- Bucala, R.; Model, P.; Cerami, A. Modification of DNA by Reducing Sugars: A Possible Mechanism for Nucleic Acid Aging and Age-Related Dysfunction in Gene Expression. Proc. Natl. Acad. Sci. USA 1984, 81, 105–109.
- Ansari, N.A.; Moinuddin, null; Mir, A.R.; Habib, S.; Alam, K.; Ali, A.; Khan, R.H. Role of Early Glycation Amadori Products of Lysine-Rich Proteins in the Production of Autoantibodies in Diabetes Type 2 Patients. Cell Biochem. Biophys. 2014, 70, 857–865.
- Olar, L.; Razvan, Ștefan; Berce, C.; Ciobanu, D.; Papuc, I. Bulletin of University of Agricultural Sciences and Veterinary Medicine Cluj-Napoca. Vet. Med. 2015, 65, 358.
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular Effects of Advanced Glycation Endproducts: Clinical Effects and Molecular Mechanisms. Mol. Metab. 2014, 3, 94–108.
- Macías-Cervantes, M.H.; Rodríguez-Soto, J.M.D.; Uribarri, J.; Díaz-Cisneros, F.J.; Cai, W.; Garay-Sevilla, M.E. Effect of an Advanced Glycation End Product-Restricted Diet and Exercise on Metabolic Parameters in Adult Overweight Men. Nutrition 2015, 31, 446–451.
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of Advanced Glycation End Products Improves Insulin Resistance in Human Type 2 Diabetes: Potential Role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616.
- Angoorani, P.; Ejtahed, H.-S.; Mirmiran, P.; Mirzaei, S.; Azizi, F. Dietary Consumption of Advanced Glycation End Products and Risk of Metabolic Syndrome. Int. J. Food Sci. Nutr. 2016, 67, 170–176.
- Saha, A.; Poojary, P.; Chan, L.; Chauhan, K.; Nadkarni, G.; DO, S.C.; Uribarri, J. Increased Odds of Metabolic Syndrome with Consumption of High Dietary Advanced Glycation End Products in Adolescents. Diabetes Metab. 2017, 43, 469–471.
- Lv, X.; Lv, G.-H.; Dai, G.-Y.; Sun, H.-M.; Xu, H.-Q. Food-Advanced Glycation End Products Aggravate the Diabetic Vascular Complications via Modulating the AGEs/RAGE Pathway. Chin. J. Nat. Med. 2016, 14, 844–855.
- Li, Z.; Wang, G.; Zhu, Y.-J.; Li, C.-G.; Tang, Y.-Z.; Jiang, Z.-H.; Yang, M.; Ni, C.-L.; Chen, L.-M.; Niu, W.-Y. The Relationship between Circulating Irisin Levels and Tissues AGE Accumulation in Type 2 Diabetes Patients. Biosci. Rep. 2017, 37.
- Chawla, D.; Bansal, S.; Banerjee, B.D.; Madhu, S.V.; Kalra, O.P.; Tripathi, A.K. Role of Advanced Glycation End Product (AGE)-Induced Receptor (RAGE) Expression in Diabetic Vascular Complications. Microvasc. Res. 2014, 95, 1–6.
- Xue, J.; Ray, R.; Singer, D.; Böhme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53, 3327–3335.
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901.
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-Glycation-End-Product-Induced Formation of Immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139.
- Gao, Z.Q.; Yang, C.; Wang, Y.Y.; Wang, P.; Chen, H.L.; Zhang, X.D.; Liu, R.; Li, W.L.; Qin, X.J.; Liang, X.; et al. RAGE Upregulation and Nuclear Factor-KappaB Activation Associated with Ageing Rat Cardiomyocyte Dysfunction. Gen. Physiol. Biophys. 2008, 27, 152–158.
- Ohashi, K.; Takahashi, H.K.; Mori, S.; Liu, K.; Wake, H.; Sadamori, H.; Matsuda, H.; Yagi, T.; Yoshino, T.; Nishibori, M.; et al. Advanced Glycation End Products Enhance Monocyte Activation during Human Mixed Lymphocyte Reaction. Clin. Immunol. 2010, 134, 345–353.
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-ΚB Pathway. Biomed. Res. Int. 2015, 2015, 732450.
- Jhun, J.; Lee, S.; Kim, H.; Her, Y.-M.; Byun, J.K.; Kim, E.-K.; Lee, S.K.; Cho, M.-L.; Choi, J.Y. HMGB1/RAGE Induces IL-17 Expression to Exaggerate Inflammation in Peripheral Blood Cells of Hepatitis B Patients. J. Transl. Med. 2015, 13.
- Bangert, A.; Andrassy, M.; Müller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Göser, S.; Korkmaz-Icöz, S.; Zittrich, S.; et al. Critical Role of RAGE and HMGB1 in Inflammatory Heart Disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164.
- Detzen, L.; Cheng, B.; Chen, C.-Y.; Papapanou, P.N.; Lalla, E. Soluble Forms of the Receptor for Advanced Glycation Endproducts (RAGE) in Periodontitis. Sci. Rep. 2019, 9, 8170.
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37.
- Schmidt, A.M. Soluble RAGEs Prospects for Treating & Tracking Metabolic and Inflammatory Disease. Vasc. Pharm. 2015, 72, 1–8.
- Farhan, S.S.; Hussain, S.A. Advanced Glycation End Products (AGEs) and Their Soluble Receptors (SRAGE) as Early Predictors of Reno-Vascular Complications in Patients with Uncontrolled Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 2457–2461.
- Gerrits, E.G.; Lutgers, H.L.; Kleefstra, N.; Graaff, R.; Groenier, K.H.; Smit, A.J.; Gans, R.O.; Bilo, H.J. Skin Autofluorescence: A Tool to Identify Type 2 Diabetic Patients at Risk for Developing Microvascular Complications. Diabetes Care 2008, 31, 517–521.
- Zerbini, G.; Maestroni, S.; Turco, V.; Secchi, A. The Eye as a Window to the Microvascular Complications of Diabetes. Dev. Ophthalmol. 2017, 60, 6–15.
- Sun, H.; Yuan, Y.; Sun, Z. Update on Mechanisms of Renal Tubule Injury Caused by Advanced Glycation End Products. Biomed. Res. Int. 2016, 2016, e5475120.
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-Induced pro-Inflammatory Responses by Retinal Müller Glia Are Regulated by the Receptor for Advanced Glycation End-Products (RAGE). Diabetologia 2010, 53, 2656–2666.
- Sato, K.; Tatsunami, R.; Yama, K.; Tampo, Y. Glycolaldehyde Induces Cytotoxicity and Increases Glutathione and Multidrug-Resistance-Associated Protein Levels in Schwann Cells. Biol. Pharm. Bull. 2013, 36, 1111–1117.
- Lu, Z.; Liu, N.; Wang, F. Epigenetic Regulations in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017.
- Espinel, E.; Agraz, I.; Ibernon, M.; Ramos, N.; Fort, J.; Serón, D. Renal Biopsy in Type 2 Diabetic Patients. J. Clin. Med. 2015, 4, 998.
- Chuang, P.Y.; Yu, Q.; Fang, W.; Uribarri, J.; He, J.C. Advanced Glycation Endproducts Induce Podocyte Apoptosis by Activation of the FOXO4 Transcription Factor. Kidney Int. 2007, 72, 965–976.
- Zhang, M.; Feng, L.; Zhu, M.; Gu, J.; Jiang, J.; Cheng, X.; Ding, S.; Wu, C.; Jia, X. The Anti-Inflammation Effect of Moutan Cortex on Advanced Glycation End Products-Induced Rat Mesangial Cells Dysfunction and High-Glucose-Fat Diet and Streptozotocin-Induced Diabetic Nephropathy Rats. J. Ethnopharmacol. 2014, 151, 591–600.
- Ki, H.-J.; Kim, S.Y.; Lee, S.H.; Moon, J.-Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Kim, S.K.; Chung, J.-H.; Kang, S.W.; et al. Transforming Growth Factor-β Receptor 2 Gene Polymorphisms Are Associated with End-Stage Renal Disease. Kidney Res. Clin. Pract. 2015, 34, 93–97.
- Miura, J.; Yamagishi, S.I.; Uchigata, Y.; Takeuchi, M.; Yamamoto, H.; Makita, Z.; Iwamoto, Y. Serum Levels of Non-Carboxymethyllysine Advanced Glycation Endproducts Are Correlated to Severity of Microvascular Complications in Patients with Type 1 Diabetes. J. Diabetes Complicat. 2003, 17, 16–21.
- Liu, J.; Huang, K.; Cai, G.-Y.; Chen, X.-M.; Yang, J.-R.; Lin, L.-R.; Yang, J.; Huo, B.-G.; Zhan, J.; He, Y.-N. Receptor for Advanced Glycation End-Products Promotes Premature Senescence of Proximal Tubular Epithelial Cells via Activation of Endoplasmic Reticulum Stress-Dependent P21 Signaling. Cell Signal 2014, 26, 110–121.
- Li, Y.; Ma, W.; Xie, C.; Zhang, M.; Yin, X.; Wang, F.; Xu, J.; Shi, B. Identification of Genes and Signaling Pathways Associated with Diabetic Neuropathy Using a Weighted Correlation Network Analysis: A Consort Study. Medicine 2016, 95, e5443.
- Araszkiewicz, A.; Gandecka, A.; Nowicki, M.; Uruska, A.; Malińska, A.; Kowalska, K.; Wierusz-Wysocka, B.; Zozulińska-Ziółkiewicz, D. Association between Small Fiber Neuropathy and Higher Skin Accumulation of Advanced Glycation End Products in Patients with Type 1 Diabetes. Pol. Arch. Med. Wewn. 2016, 126, 847–853.
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced Glycation End Products in Extracellular Matrix Proteins Contribute to the Failure of Sensory Nerve Regeneration in Diabetes. Diabetes 2009, 58, 2893–2903.
- Loske, C.; Neumann, A.; Cunningham, A.M.; Nichol, K.; Schinzel, R.; Riederer, P.; Münch, G. Cytotoxicity of Advanced Glycation Endproducts Is Mediated by Oxidative Stress. J. Neur. Transm 1998, 105, 1005–1015.
- Yu, T.; Li, L.; Chen, T.; Liu, Z.; Liu, H.; Li, Z. Erythropoietin Attenuates Advanced Glycation Endproducts-Induced Toxicity of Schwann Cells in Vitro. Neurochem. Res. 2015, 40, 698–712.
- Guitart, K.; Loers, G.; Schachner, M.; Kleene, R. Prion Protein Regulates Glutathione Metabolism and Neural Glutamate and Cysteine Uptake via Excitatory Amino Acid Transporter 3. J. Neurochem. 2015, 133, 558–571.
- Bus, S.A.; Haspels, R.; Busch-Westbroek, T.E. Evaluation and Optimization of Therapeutic Footwear for Neuropathic Diabetic Foot Patients Using In-Shoe Plantar Pressure Analysis. Diabetes Care 2011, 34, 1595–1600.
- Vouillarmet, J.; Maucort-Boulch, D.; Michon, P.; Thivolet, C. Advanced Glycation End Products Assessed by Skin Autofluorescence: A New Marker of Diabetic Foot Ulceration. Diabetes Technol. 2013, 15, 601–605.
- American Diabetes Association. Screening Guidelines for Diabetic Retinopathy: Clinical Guideline. Ophthalmology 1992, 99, 1626–1628.
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58.
- Tracey, M.L.; McHugh, S.M.; Fitzgerald, A.P.; Buckley, C.M.; Canavan, R.J.; Kearney, P.M. Trends in Blindness Due to Diabetic Retinopathy among Adults Aged 18-69years over a Decade in Ireland. Diabetes Res. Clin. Pract. 2016, 121, 1–8.
- Kowluru, R.A. Effect of Advanced Glycation End Products on Accelerated Apoptosis of Retinal Capillary Cells under in Vitro Conditions. Life Sci. 2005, 76, 1051–1060.
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424.
- Cheng, L.; Bu, H.; Portillo, J.-A.C.; Li, Y.; Subauste, C.S.; Huang, S.S.; Kern, T.S.; Lin, F. Modulation of Retinal Müller Cells by Complement Receptor C5aR. Invest. Ophthalmol. Vis. Sci. 2013, 54, 8191–8198.
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Sato, T.; Takeuchi, M. Potential Utility of Statins, 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors in Diabetic Retinopathy. Med. Hypotheses 2006, 66, 1019–1021.
- AI, J.; LIU, Y.; SUN, J.-H. Advanced Glycation End-Products Stimulate Basic Fibroblast Growth Factor Expression in Cultured Müller Cells. Mol. Med. Rep. 2013, 7, 16–20.
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced Glycation End-Products Induce Basement Membrane Hypertrophy in Endoneurial Microvessels and Disrupt the Blood-Nerve Barrier by Stimulating the Release of TGF-β and Vascular Endothelial Growth Factor (VEGF) by Pericytes. Diabetologia 2011, 54, 1517–1526.
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New Type of Cardiomyopathy Associated with Diabetic Glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602.
- Abd-El Aziz, F.M.; Abdelghaffar, S.; Hussien, E.M.; Fattouh, A.M. Evaluation of Cardiac Functions in Children and Adolescents with Type 1 Diabetes. J. Cardiovasc. Ultrasound. 2017, 25, 12–19.
- Yang, Q.; Gao, H.; Dong, R.; Wu, Y.-Q. Sequential Changes of Endoplasmic Reticulum Stress and Apoptosis in Myocardial Fibrosis of Diabetes Mellitus-Induced Rats. Mol. Med. Rep. 2016, 13, 5037–5044.
- Novoa, U.; Arauna, D.; Moran, M.; Nuñez, M.; Zagmutt, S.; Saldivia, S.; Valdes, C.; Villaseñor, J.; Zambrano, C.G.; Gonzalez, D.R. High-Intensity Exercise Reduces Cardiac Fibrosis and Hypertrophy but Does Not Restore the Nitroso-Redox Imbalance in Diabetic Cardiomyopathy. Oxid. Med. Cell Longev. 2017, 2017, 7921363.
- Cao, W.; Chen, J.; Chen, Y.; Chen, X.; Liu, P. Advanced Glycation End Products Promote Heart Failure through Inducing the Immune Maturation of Dendritic Cells. Appl. Biochem. Biotechnol. 2014, 172, 4062–4077.
- Zerif, E.; Maalem, A.; Gaudreau, S.; Guindi, C.; Ramzan, M.; Véroneau, S.; Gris, D.; Stankova, J.; Rola-Pleszczynski, M.; Mourad, W.; et al. Constitutively Active Stat5b Signaling Confers Tolerogenic Functions to Dendritic Cells of NOD Mice and Halts Diabetes Progression. J. Autoimmun. 2017, 76, 63–74.
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory Role of Dendritic Cells in Postinfarction Healing and Left Ventricular Remodeling. Circulation 2012, 125, 1234–1245.
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A Beta Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006.
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated Collagen Cross-Linking Alters Cardiac Mechanics in Volume-Overload Hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1277–H1284.
- Willemsen, S.; Hartog, J.W.L.; Hummel, Y.M.; van Ruijven, M.H.I.; van der Horst, I.C.C.; van Veldhuisen, D.J.; Voors, A.A. Tissue Advanced Glycation End Products Are Associated with Diastolic Function and Aerobic Exercise Capacity in Diabetic Heart Failure Patients. Eur. J. Heart Fail 2011, 13, 76–82.
- Fang, M.; Wang, J.; Li, S.; Guo, Y. Advanced Glycation End-Products Accelerate the Cardiac Aging Process through the Receptor for Advanced Glycation End-Products/Transforming Growth Factor-β-Smad Signaling Pathway in Cardiac Fibroblasts. Geriatr. Gerontol. Int. 2016, 16, 522–527.
- Kawashima, T.; Inuzuka, Y.; Okuda, J.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Kawamoto, A.; Narazaki, M.; Matsuda, T.; et al. Constitutive SIRT1 Overexpression Impairs Mitochondria and Reduces Cardiac Function in Mice. J. Mol. Cell Cardiol. 2011, 51, 1026–1036.
- Gu, X.S.; Wang, Z.B.; Ye, Z.; Lei, J.P.; Li, L.; Su, D.F.; Zheng, X. Resveratrol, an Activator of SIRT1, Upregulates AMPK and Improves Cardiac Function in Heart Failure. Genet. Mol. Res. 2014, 13, 323–335.
- Yuan, Q.; Zhou, Q.-Y.; Liu, D.; Yu, L.; Zhan, L.; Li, X.-J.; Peng, H.-Y.; Zhang, X.-L.; Yuan, X.-C. Advanced Glycation End-Products Impair Na+/K+-ATPase Activity in Diabetic Cardiomyopathy: Role of the Adenosine Monophosphate-Activated Protein Kinase/Sirtuin 1 Pathway. Clin. Exp. Pharm. Physiol. 2014, 41, 127–133.
- Yan, D.; Luo, X.; Li, Y.; Liu, W.; Deng, J.; Zheng, N.; Gao, K.; Huang, Q.; Liu, J. Effects of Advanced Glycation End Products on Calcium Handling in Cardiomyocytes. CRD 2014, 129, 75–83.
- Niggli, E. The Cardiac Sarcoplasmic Reticulum. Circ. Res. 2007, 100, 5–6.
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Förster, A.; Schmitto, J.D.; Gummert, J.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II and Protein Kinase A Differentially Regulate Sarcoplasmic Reticulum Ca2+ Leak in Human Cardiac Pathology. Circulation 2013, 128, 970–981.


