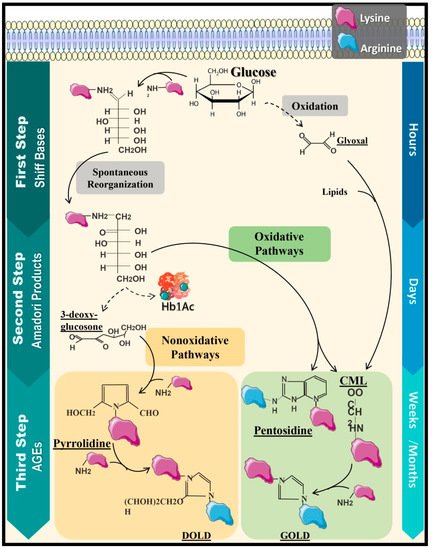
Chronic complications of DM derive from structural and functional modifications of blood vessels due to hyperglycemia, affecting the heart, kidneys, and nervous system
[27]. Among the mechanisms implied in developing these complications are the structural modifications induced by AGEs in vulnerable molecules such as proteins, lipids, and DNA, altering their stability and functions
[28].
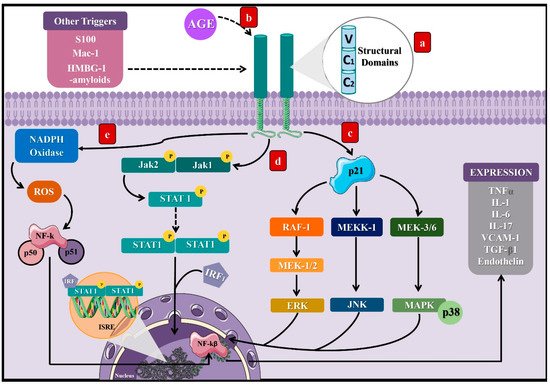
Activation of the receptor for advanced glycation end products (RAGE) represents the main mechanism involved between DM and AGEs
[8]. RAGE is a cell surface receptor composed of three extracellular domains, a transmembrane domain, and a cytoplasmic tail
[8], which belongs to the immunoglobulin superfamily
[29], since its expression derives from the major histocompatibility complex class III (MHC-III)
[30]. Thus, RAGE expression predominates in specific cells such as monocytes, macrophages, proximal tubular cells, podocytes, and mesangial cells
[29].
As a result of the AGE–RAGE interaction, the cytoplasmic domain of RAGE leads to different signaling pathways. In particular, it can activate the p21 protein
[31], triggering other signaling compounds to eventually stimulate kinases such as the extracellular signal-regulated kinase (ERK), c-Jun
N-terminal kinase (JNK), and mitogen-activated protein kinase (MAP)
[29], along with Janus kinase 1 and 2/Signal transducer and activators of transcription (JAK/STAT1)
[32]. Finally, the consequences of these signal transduction pathways consist of the activation of transcription factors such as nuclear factor kappa B (NF-kB)
[33] and the interferon-sensitive response element (ISRE)
[34], which lead to the synthesis of proinflammatory cytokines as tumor necrosis factor-alpha (TNF-α)
[34] and interleukins (IL) 1, 6, and 17
[35][36][35,36], as well as vascular cell adhesion molecule-1 (VCAM-1)
[29].
Additionally, NADH oxidase’s activation directly and indirectly generates reactive oxygen species (ROS) due to RAGE stimulation
[37]. Furthermore, RAGE can be activated by other types of ligands aside from AGEs, including S 100 or Calgranulin, Mac-1, High-mobility group box 1 (HMBG1), and β-amyloids, predominantly when their levels increase during inflammatory reactions (
Figure 2)
[31]. The discovery of RAGE’s multiligand nature explains its elevated and persistent activity in diabetes complications
[36][37][36,37].
Figure 2. Signaling pathways induced by the activation of the receptor of AGEs. (a) RAGE is a cell surface receptor composed of three extracellular domains, a transmembrane domain, and a short cytoplasmic tail. (b) AGEs bind to RAGE’s extracellular portion and induce activation of the cytoplasmic domain, leading to different signaling pathways, which will finally result in the stimulation of transcription factors such as NF-KB and ISRE. Three pathways that can lead to such a response: (c) activation of the protein p21, which induces RAF-1, MEKK-1, and MEK 3/6 proteins that activate the factors ERK, JNK, and MAPK, which translocate to the cell nucleus. (d) Activation of the JAK2/STAT1 pathway, where STAT1 dimerizes and binds to the IRF1 sequence to translocate to the cell nucleus, binding to the ISRE segment and inducing transcription of proinflammatory cytokines. (e) Activation of the NADPH oxidase that leads to the stimulation of NF-KB. These processes can be activated by other molecules such as S100, Mac-1, HMBG-1, and β-amyloids. NF-kB: nuclear factor kappa light chain enhancer of activated B cells, ISRE: interferon-sensitive response element, TNFα: tumor necrosis factor-alpha, IL: interleukins, VCAM-1: adherence and growth factors as the vascular cell adhesion molecule-1, TGF-β1: transforming growth factor β, RAF-1: proto-oncogene serine/threonine kinase, MEKK-1: mitogen-activated protein kinase kinase kinase 1, MEK 3/6: mitogen-activated protein kinase kinase, ERK: extracellular signal-regulated kinase, JNK: c-Jun N-terminal kinase, MAPK: mitogen-activated protein kinase, JAK: Janus kinase, STAT: signal transducer and activators of transcription, IRF1: interferon regulatory factor-1, S 100: calgranulin, and HMBG1: high-mobility group box 1.
There are different types of these receptors that have been studied, known as soluble forms of RAGE (RAGEs), which are composed of extracellular domains without their intracellular portion, so they can be transported and found free in the plasma
[38]. Of note, two types of receptors have been identified, cleaved RAGE(cRAGE) and endogenous secretory RAGE (esRAGE) or RAGE_V1, depending on how they were created
[39]. cRAGE is formed from the action of matrix metalloproteases (MMP) and α-disintegrin metalloprotease (ADAM)-10; these enzymes cleave from the cell surface to RAGE, thus losing its transmembrane and cytosolic portions, but it conserves the V1-C1-C2 domains of RAGE. On the other hand, esRAGE is generated by the alternative splicing of the RNAm RAGE gene, changing its structure by adding a 16-amino acid extension at the c-terminal end
[40].
Although the distribution and function of these receptors are not yet clear, there are multiple hypotheses about their role in the pathophysiology of inflammatory and metabolic diseases, such as DM. Among the most managed actions, their role as “decoys” for AGEs is evaluated, generating a downregulation effect in inflammation and preventing cell damage due to the sequestration of RAGE ligands that leads to the pathways not activated being intracellularly related to these receptors not only by AGEs but other ligands such as HMB1 or S100, postulating a possible regulatory action on the AGEs–RAGE axis
[40]. In addition, studies have established an inversely proportional relationship between the levels of sRAGE and the markers of metabolic syndrome and atherosclerosis in patients with DM, used as a biomarker in inflammatory processes and complications associated with this disease
[41].
3.1. Molecular Mechanisms of AGEs in Microvascular Complications of DM
The presence of diabetic retinopathy, neuropathy, or (micro) albuminuria defines the existence of microvascular complications of DM
[42]; despite affecting different organs, these complications mutually relate to each other
[43]. Diverse studies have associated AGEs with the progression of those complications, mainly given the direct action of these products on tissues or via stimulation of the AGE–RAGE axis and the subsequent inflammatory response
[44][45][46][44,45,46].
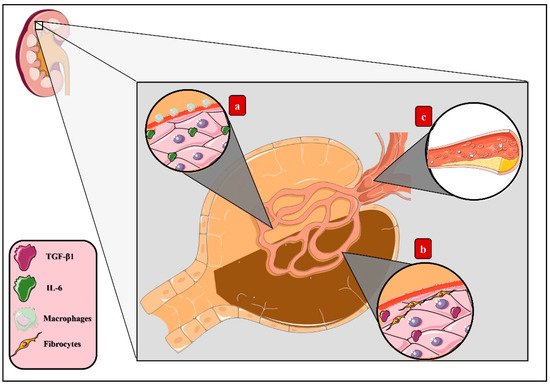
Diabetic kidney disease (DKD) is characterized by renal hypertrophy, proteinuria, decreased glomerular filtration rate, and renal fibrosis
[47], ultimately progressing to chronic kidney disease (CDK)
[48]. Induction of the AGE–RAGE pathways deriving from the accumulation of these products in renal tissue leads to inflammatory activity
[49]. Thus, triggering the migration of macrophages that agglomerate in the renal glomerulus’s mesangium and establishing an inflammatory microenvironment led by IL-6 synthesis with the consequent expansion of this layer eventually causes the compression of capillaries and reduction of the body surface area of renal filtration
[50]. Additionally, increased expression of transforming growth factor β (TGF-β) has been correlated with fibrogenesis activation, collagen synthesis stimulation, and renal tubular cell apoptosis, therefore explaining both glomerular sclerosis and dysfunction
[51].
Other mechanisms induced by the AGE–RAGE pathway arise specifically through CML, which constitutes a higher AGE accumulation in vivo
[52], the renal epithelium being continuously exposed to these changes
[53]. It has been recently determined that AGEs generate lipid accumulation in this tissue deriving from altered cholesterol metabolism, since AGEs activate the sterol-regulatory element-binding protein 2 (SREBP-2) and, correspondingly, the expression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) increases, concluding in an increased cholesterol synthesis. Additionally, this molecule’s access to cells benefits from the stimulation of low-density lipoprotein (LDLc) activity in conjunction with the decrease of the ATP-binding cassette transporter A1 (ABCA1), which results in tissue dysfunction
[44] (
Figure 3).
Figure 3. AGEs in diabetic kidney disease. Activation of the AGE–RAGE axis derived from the accumulation of AGEs in renal tissue induces tissue dysfunction through diverse mechanisms: (a) macrophage migration, which agglomerates in the renal glomerulus’s mesangium, establishing an inflammatory microenvironment led by IL-6 synthesis and, eventually, causing the expansion of this layer, compression of the capillary, and reduction of the body surface area of filtration. (b) Increased expression of transforming growth factor β (TGF-β), which stimulates fibrogenesis, collagen synthesis, and renal tubular cell apoptosis, leading to glomerular sclerosis. (c) Lipids storage from altered cholesterol metabolism as a result of the activation of sterol-regulatory element-binding protein 2 (SREBP-2), increasing the expression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) and, finally, concluding in increased cholesterol synthesis.
Diabetic neuropathy is defined by the progressive loss of axons within peripheral nerves, clinically manifested by severe pain and sensory impairment
[54]. The accumulation of AGEs in the endoneurium, Schwann cells, extracellular matrix, and capillary within these nervous structures cause the glycation of proteins such as fibronectin and laminin
[55], inducing structural and functional modifications that decrease the regenerative capacity related to axonal atrophy
[56]. Likewise, oxidative stress and, thereby, neuronal cytotoxicity are induced through the AGE–RAGE pathway
[57], given the increased levels of superoxide and hydrogen peroxide
[58] and decreased intracellular glutathione (GSH)
[46], which is an essential antioxidant tripeptide composed of glutamate, cysteine, and glycine
[59].
The loss of peripheral sensation and the increase of mechanical pressure in the feet are the primary cause of diabetic foot
[60]. Secondly, the oxidative stress, proinflammatory cytokines presence, and glycation of proteins such as collagen lead to the hardening of epithelial cells’ basement membranes, concluding in skin tissue frailty and impaired wound healing
[61].
On the other hand, diabetic retinopathy (DR) constitutes a degenerative vascular process that progresses through different stages
[62]. First, a blood flow imbalance emerges, in addition to an increased vascular permeability and capillary basement membrane hardening, advancing to the formation of microaneurysms and establishing a microvascular injury that produces ischemia due to decreased retinal blood flow, thus representing a significant cause of blindness
[63][64][63,64]. The development of these pathological changes results from pericyte apoptosis induced by the AGE–RAGE pathway. Likewise, increased oxidative stress produced by NF-kB expression produces free radicals such as peroxynitrite inside the subretinal membrane and microvasculature, damaging the DNA
[65].
Moreover, the regulatory function of Müller cells inside the retina
[66] becomes affected in DM by exposure to hyperglycemia
[67], and the inflammatory process, alongside its effects on the microvasculature, is also a consequence of activation of the AGE–RAGE pathway
[45]. Furthermore, this pathway increases the expression of cytokines and proangiogenic factors such as the vascular endothelial growth factor (VEGF)
[68], basic fibroblast growth factor (bFGF)
[69], and TGF-β
[70], leading to the distinguished neovascularization of DR and significantly exacerbated by the high accumulation capacity of AGEs in the vitreous humor
[69].
3.2. AGEs and the Macrovascular Alterations in DM
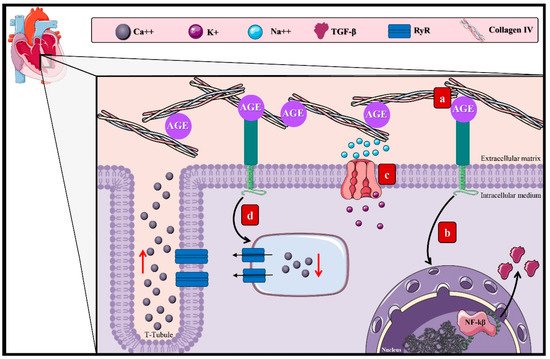
Cardiovascular complications of DM arise as a consequence of the damage to large-diameter vascular structures. They are mostly the leading cause of death among diabetic patients, representing 50% of the deaths related to this disease
[27]. Diabetic cardiomyopathy is characterized by ventricular dysfunction originating from myocyte hypertrophy
[71][72][71,72] and myocardial fibrosis
[73]. The AGE–RAGE axis has been admitted as one of the contributing factors to this incompletely elucidated chronic complication (
Figure 4)
[29].
Figure 4. AGEs in diabetic cardiomyopathy. The effect of the AGEs in diabetic cardiomyopathy arises through diverse mechanisms: (a) accumulation in the extracellular matrix of cardiac tissue due to the interaction with structural proteins, inducing reticulation between collagen fibers and laminin and decreasing the elastic properties of the cardiac tissue. (b) Activation of the AGE–RAGE pathway by the induction of TGF-β and other proinflammatory cytokines that allow the proliferation of fibroblasts conducive to myocardial hypertrophy. (c) Activation of the AGE–RAGE pathway and ionic imbalance due to inhibition of the SIRT1/NAD+ pathway, disturbing the function of the Na++/K+ ATPase. (d) Overstimulation of the RyR, generating irregular modifications of the Ca++ levels, favoring its exit and causing an alteration of the cardiac cycle that advances to diastolic dysfunction. Ca++: calcium, K+: potassium, Na++: sodium, TGF-β1: transforming growth factor β, and RyR: ryanodine receptors.
This process may cause the deterioration of cardiac functions by myocyte hypertrophy
[74]. Lately, it has been established that this cardiac remodeling process occurs through the connection within the AGE–RAGE pathway and dendritic cells (DC)
[75], which are antigen-presenting cells with essential functions in T-cell regulation and homeostasis
[76]. However, it has been reported that the accumulation of matured DC during myocardial infarction could aggravate the tissue remodel
[77]. Equally, during in vitro studies, it was determined that the AGE–RAGE pathway promotes DC’s maturation and, therefore, the expression of genes that develop hypertrophy, such as
MYH7, which encodes the cardiac beta-myosin heavy chain (β-MHC)
[75][78][75,78].
The increment in fibroblast numbers after the increase of AGEs in the extracellular matrix
[74] promotes interactions with structural proteins, inducing reticulation between collagen fibers and laminin, deriving in a loss of the cardiac tissue’s elastic properties, rigidity, and increased cardiac volume, conductive to diastolic dysfunction
[79][80][79,80]. The AGE–RAGE pathway intervenes in fibroblast proliferation by stimulating proinflammatory genes and TGF-β, amplifying the adverse effect on the cardiac elastic properties
[81].
Separately, the accumulation of AGEs in cardiac tissue is also related to the inhibition of the sirtuin-1 protein (SIRT1) expression. SIRT1, a member of the class III deacetylase family, is an antioxidant protein capable of delaying fibrosis and apoptosis of cardiac cells through its activation by NAD
+ [82]. Besides, the adenosine monophosphate-activated protein kinase (AMPK) keeps a cellular energetic balance and enhances the NAD
+ levels and can also regulate SIRT1 functions
[83]. In conclusion, it has been established that Na
++/K
+ ATPase alterations are due to dysregulation of the SIRT1/AMPK pathway, modifying cellular ionic homeostasis
[84].
Moreover, the Ca
2+ levels decrease due to the increased activity of the ryanodine receptors induced by AGE–RAGE
[85]. These receptors manage to equilibrate the ion levels during diastole and systole
[86]; however, their hyperactivity allows a Ca
2+ leak from the sarcoplasmic reticulum during diastole, diminishing the Ca
2+ levels during systole and, thus, disturbing the cardiac cycle
[87], driving to cardiac dysfunction
[85].