Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stijn Wenmaekers | + 1750 word(s) | 1750 | 2021-05-25 11:28:32 | | | |
| 2 | Catherine Yang | Meta information modification | 1750 | 2021-07-19 12:08:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wenmaekers, S. HUWE1. Encyclopedia. Available online: https://encyclopedia.pub/entry/12182 (accessed on 07 February 2026).
Wenmaekers S. HUWE1. Encyclopedia. Available at: https://encyclopedia.pub/entry/12182. Accessed February 07, 2026.
Wenmaekers, Stijn. "HUWE1" Encyclopedia, https://encyclopedia.pub/entry/12182 (accessed February 07, 2026).
Wenmaekers, S. (2021, July 19). HUWE1. In Encyclopedia. https://encyclopedia.pub/entry/12182
Wenmaekers, Stijn. "HUWE1." Encyclopedia. Web. 19 July, 2021.
Copy Citation
HUWE1 is postulated as a therapeutic response modulator, affecting the collision between platinum-DNA adducts and the replication fork, the primary cytotoxic action of platins.
chemotherapy
resistance
biomarkers
translational medicine research
1. Introduction
HUWE1 is a ubiquitin (Ub) E3 ligase that functions as a terminating enzyme in the process of protein ubiquitination. Following the consecutive actions of Ub-activating (E1) and -conjugating (E2) enzymes, HUWE1 post-translationally modifies other proteins by adding different types of Ub chains. A well-known function of HUWE1 is that it targets other proteins for degradation in the Ub-proteasome system (UPS) via K48-linked poly-Ub chains [1]. In contrast to being a facilitator of protein degradation, HUWE1 regulates processes such as protein activation and cellular signal transduction via K63-linked polyubiquination [1][2]. HUWE1-mediated monoubiquitination and less well understood K6-linked polyubiquitination further indicate its multifaceted cellular regulatory effects [3][4][5][6]. Deubiquitinating enzymes on the other hand counteract the post-translational actions of HUWE1. In addition to the catalytic HECT domain, HUWE1 contains a Ub-associated (UBA) domain, a Ub-binding motif (UBM1) domain, and a Bcl-2 homology region 3 (BH3) domain [7][8]. While the functions of the UBM1- and UBA domains are still obscure, evidence indicates that the BH3 domain allows HUWE1 to specifically interact with the induced myeloid leukemia cell differentiation protein (Mcl-1), an anti-apoptotic member of the B-cell lymphoma 2 (Bcl-2) family [9]. This interaction implies a modulatory role for HUWE1 in apoptosis. Many other HUWE1 targets with roles in the DDR have already been identified [10].
HUWE1-mediated ubiquitination is a highly coordinated process and deregulation has been linked to tumorigenesis as well as tumor suppression [7][8]. Given its modulatory role on mediators such as Mcl-1, the breast cancer type 1 susceptibility protein (BRCA1) and p53, HUWE1 is linked to DNA damage repair pathways and apoptosis [9][11][12]. These processes are not only hallmarks of cancer, but also influence cellular sensitivity to platins and other genotoxins [13][14].
2. HUWE1 Modulates the Intrinsic Apoptotic Pathway
A severely threatened genome shifts the DDR from a reparative state towards controlled cell death [15]. Apoptotic signaling follows interconnected patterns that ultimately converge on caspase-executioners for cellular decay. Genotoxins including platins rely on the intrinsic, mitochondrial, pathway to induce cell death. Intrinsic apoptosis is regulated by a delicate balance between pro- and anti-apoptotic members of the Bcl-2 family. These control the release of cytochrome c (cyt c) into the cytoplasm [16][17]. In response to genotoxic stress, pro-apoptotic Bcl-2 induction destabilizes the mitochondrial membrane, thereby facilitating the cytoplasmatic release of cyt c and caspase activation [17]. The tumor suppressor p53 is a principal pro-apoptotic non-Bcl-2 protein and DDR effector that promotes this process [18]. Post-translational activation of p53 in response to cellular stress triggers an apoptotic cascade [19].
The intrinsic apoptotic pathway is influenced by the effects of HUWE1. Considering this process, HUWE1 directly regulates p53 and Mcl-1, an anti-apoptotic member of the Bcl-2 family. In addition, HUWE1 impairs the post-translational activation of p53 via regulation of ATM and histone deacetylase 2 (HDAC2) (Figure 1). HUWE1-mediated effects on the cellular ability to induce apoptosis in response to platins might be considered as a potent driver of therapy sensitivity.
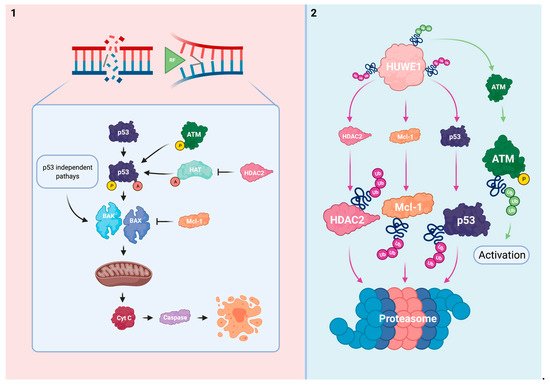
Figure 1. HUWE1 influences the intrinsic apoptotic pathway. (1) In response to irreparable DNA damage, p53 is activated via ATM-mediated phosphorylation and HAT-mediated acetylation. HDAC2 inhibits acetylation of p53. The pro-apoptotic BAK/BAX are stimulated by p53 activation and via p53 independent mechanisms. This subsequently results in mitochondrial pore formation and the cytoplasmatic release of cyt c to effectuate apoptosis. The anti-apoptotic Mcl-1 inhibits BAK/BAX. (2) HUWE1 negatively regulates p53, Mcl-1, and HDAC2 via polyubiquitination. HUWE1 positively regulates ATM phosphorylation via polyubiquitination. HUWE1 depletion could result in cellular immortalization and resistance to platins via target overexpression and subsequent ineffective activation of the intrinsic apoptotic pathway.
P53 loss of function occurs frequently in various cancers and can lead to cellular immortalization [20]. Normally, cells maintain p53 at low levels to maintain their proliferative capabilities via mechanisms such as the mouse double minute 2 homolog (MDM2), an E3 Ub ligase. However, in response to cellular stress, p53 is rapidly phosphorylated and stabilized to block MDM2-mediated degradation [21]. In addition to phosphorylation, histone acetyl-transferase (HAT)-mediated p53 acetylation is indispensable for its activation [22]. Activated p53 stimulates multiple proteins including the Bcl-2 homologous antagonist/killer (BAK) and Bcl-2 associated X (BAX) to shift cellular balance towards a pro-apoptotic state [23][24]. However, BAK and BAX can also be activated in p53 deficient conditions due to Mcl-1 neutralization, thereby indicating the relevance of the interplay between HUWE1 and intrinsic apoptosis even in a p53 mutant tumor [25].
HUWE1 has been found to directly target p53 for proteasomal degradation in multiple models [10][12][26][27]. This reciprocal relationship at steady state seems to promote the cell’s proliferative capabilities similarly to the MDM2/p53 complex. Conversely, a positive relationship between HUWE1 and p53 has been identified in multiple thyroid cancer models. HUWE1 overexpression increased p53 stability by MDM2 downregulation in thyroid cancer (WRO) cells and mouse xenografts. Ectopic HUWE1 expression in HUWE1 KD thyroid cancer cells sensitized this model to cisplatin and other genotoxins [28].
ATM activation is mediated by HUWE1 in B-cells and MEFs as previously discussed. In HUWE1 deficient and p53 sufficient conditions, these cells failed to successfully trigger a p53 response cascade upon exposure to doxorubicin, etoposide, and y-irradiation due to insufficient levels of activated ATM. Interestingly, the response to dexamethasone, a p53-independent inducer of apoptosis, was not influenced by HUWE1 status in B-cells [26].
HDAC2 has been identified as a HUWE1 target for proteasomal degradation in MEFs [29]. HUWE1 deficiency impaired the ability to induce apoptosis in response to cisplatin or nutlin-3 treatment via HDAC2 enhancement in these cells. Whereas phosphorylated and acetylated p53 accumulated in HUWE1 WT-expressing controls, HUWE1 KO MEFs failed to effectuate these post-translational modifications upon genotoxic stress. Subsequently, these cells could not effectively initiate a downstream p53 response cascade. Reduction of HDAC2 to near WT levels was sufficient to normalize the stress-induced p53 cascade and re-sensitized HUWE1 KO MEFs to cisplatin and nutlin-3 treatment [29]. Taken together, these data imply that loss of HUWE1 impairs the ability to effectively trigger and effectuate p53-mediated apoptosis in response to platins and other genotoxins.
Mcl-1 exerts its anti-apoptotic function primarily by complex formation with the pro-apoptotic BAK and BAX proteins via their shared BH3 domains. Within these complexes BAK and BAX’s ability to destabilize the mitochondrial membrane is impaired [30].
HUWE1′s BH3 domain mostly resembles BAK’s BH3 domain allowing it to act as a dose-dependent regulator of Mcl-1 in HeLa cells [9][31]. Whereas the BH3 domain serves as a Mcl-1 docking station, the consecutive action of HUWE1′s catalytic HECT domain ubiquitinates Mcl-1 for proteasomal degradation [9]. Cellular Mcl-1 has been reported to accumulate in HUWE1 deficient conditions [9][29][31][32][33]. Interaction between HUWE1 and other Bcl-2 family members was not observed in HeLa cells [9][31]. However, HUWE1 KD in an ischemic cortical neuron model modulated Bcl-2 and BAX additionally to Mcl-1 which further promoted the anti-apoptotic phenotype [34]. HUWE1 might thus have a more comprehensive role in regulation of Bcl-2 members.
The direct relationship between HUWE1, Mcl-1, and the response to platins has been described in several reports [9][29][32][35]. HUWE1 KD in HeLa cells impaired the apoptotic response upon UV-irradiation, etoposide, or cisplatin exposure via Mcl-1 enhancement. Of the investigated genotoxic agents, cisplatin proved to be the least potent to trigger apoptosis [9]. In line with these findings, Mcl-1 reduction to WT levels re-sensitized HUWE1 KO MEFs to cisplatin treatment [29]. Others indicated that bile salt-induced Mcl-1 phosphorylation enhanced Mcl-1 stability by blocking HUWE1-mediated degradation in human liver cancer (HepG2) cells. The subsequent inability to effectuate apoptosis upon cisplatin treatment could be rescued by Mcl-1 KD in these cells [35]. Similarly to the observations in cell models, intestinal crypts of transgenic colorectal cancer HUWE1 KO mice displayed a decreased cisplatin sensitivity by Mcl-1 upregulation [32]. HUWE1 deficiency thus protects cells from undergoing apoptosis in response to platins and other genotoxins via Mcl-1 upregulation.
Mcl-1 is considered one of HUWE1′s principal targets in oncology. Next to HUWE1, Mcl-1 turnover is also mediated by the β-transducin repeat-containing E3 Ub protein ligase and Ub-independent pathways [36][37]. In addition to the direct evidence between the HUWE1/Mcl-1 axis and cisplatin sensitivity, Mcl-1 overexpression is a well-known inducer of resistance to platins and other genotoxins [38][39]. The potential anti-neoplastic use of Mcl-1 inhibitors is currently being investigated [40].
Non-functional mutagenic p53 can impair the apoptotic response and is linked to platinum-based therapy resistance [20][41]. However, the exact role of p53 in modulating sensitivity to platins is complex and cell context-dependent [42]. Mutagenic loss of p53 function is accompanied by conformational changes that influence its susceptibility to post-translational modifications for protein activation. Interestingly, the impaired ability to acetylate mutated p53 is linked to its decreased functionality [43]. In addition, overexpression and cytoplasmatic mislocalization of p53 have been linked to platinum-based therapy resistance by inhibiting caspase effectors [44]. Similarly to mutagenic p53, oncogenic loss of HUWE1 might decrease the cellular responsiveness to platins via impaired p53 activation. However, the reported relationship between HUWE1 and p53 varies among cell types and depends on the interplay with MDM2 [10][12][26][27][28]. This strengthens the concept that Ub network organization varies in spatiotemporal dimensions [45]. Whereas inhibition of MDM2 with nutlin-3 sensitizes cells to platins, this mechanism of action relies on enhancing p53 levels rather than promoting its post-translational activation [46][47]. Nonetheless, it highlights the potential of targeting UPS mediators to promote p53-mediated apoptosis. Complementary to its principal role in apoptotic signaling, one should note that p53 has a broad spectrum of other effects within the DDR [48].
Next to the direct evidence on HDAC2 as a cisplatin desensitizer upon loss of HUWE1, HDAC2-induced resistance to cisplatin and other DNA damaging agents is supported within other conditions [49][50]. Other HDACs are however involved in the post-translational modification of p53 which further indicates the potential existence of cellular rescue and compensation mechanisms [51]. In addition to its non-histone effects on p53, HDAC2-mediated deacetylation of histones results in DNA condensation [52]. This impairs the DNA accessibility for platins to induce DNA damage [53]. The use of HDAC inhibitors is an emerging field of cancer drug research that could be especially promising when applied synergistically with established cancer drugs such as platins [54][55].
Platinum’s primary mode of action is inseparably connected to the intrinsic apoptotic pathway. Altogether, the direct and indirect regulatory effects of HUWE1 on Mcl-1 and p53 propose an important role for HUWE1 in successfully triggering intrinsic apoptosis in response to platins. This suggests that HUWE1 has the potential to act as a biomarker to assess an individual’s response to platins. Moreover, stimulation of HUWE1 could induce the intrinsic apoptotic pathway which further promotes HUWE1 as a sensitizer of platinum-based therapy.
References
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J. 2020, 287, 1985–1999.
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell 2005, 123, 409–421.
- Parsons, J.; Tait, P.S.; Finch, D.; Dianova, I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215.
- Choe, K.N.; Nicolae, C.M.; Constantin, D.; Kawasawa, Y.I.; Delgado-Diaz, M.R.; De, S.; Freire, R.; Smits, V.A.; Moldovan, G. HUWE 1 interacts with PCNA to alleviate replication stress. EMBO Rep. 2016, 17, 874–886.
- Markkanen, E.; Van Loon, B.; Ferrari, E.; Parsons, J.; Dianov, G.L.; Hubscher, U. Regulation of oxidative DNA damage repair by DNA polymerase and MutYH by cross-talk of phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2012, 109, 437–442.
- Michel, M.A.; Swatek, K.N.; Hospenthal, M.K.; Komander, D. Ubiquitin Linkage-Specific Affimers Reveal Insights into K6-Linked Ubiquitin Signaling. Mol. Cell 2017, 68, 233–246.e5.
- Gong, X.; Du, D.; Deng, Y.; Zhou, Y.; Sun, L.; Yuan, S. The structure and regulation of the E3 ubiquitin ligase HUWE1 and its biological functions in cancer. Investig. New Drugs 2020, 38, 515–524.
- Kao, S.-H.; Wu, H.-T.; Wu, K.-J. Ubiquitination by HUWE1 in tumorigenesis and beyond. J. Biomed. Sci. 2018, 25, 1–15.
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell 2005, 121, 1085–1095.
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule Is a Critical Mediator of the ARF Tumor Suppressor. Cell 2005, 121, 1071–1083.
- Wang, X.; Lu, G.; Li, L.; Yi, J.; Yan, K.; Wang, Y.; Zhu, B.; Kuang, J.; Lin, M.; Zhang, S.; et al. HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin–proteasome pathway. Biochem. Biophys. Res. Commun. 2014, 444, 549–554.
- Yang, D.; Cheng, D.; Tu, Q.; Yang, H.; Sun, B.; Yan, L.; Dai, H.; Luo, J.; Mao, B.; Cao, Y.; et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018, 8, 3517–3529.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103.
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248.
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883.
- Henkels, K.M.; Turchi, J.J. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999, 59, 3077–3083.
- Han, J.Y.; Chung, Y.J.; Kim, J.S.; Rhyu, M.G.; Kim, H.K.; Lee, K.S.; Park, S.W. The relationship between cisplatin—induced apoptosis and p53, bcl-2 and bax expression in human lung cancer cells. Korean J. Intern. Med. 1999, 14, 42–52.
- Brooks, C.L.; Gu, W. New insights into p53 activation. Cell Res. 2010, 20, 614–621.
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M.; et al. p53 Mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839.
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334.
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626.
- Leu, J.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D. Mitochondrial p53 activates BAK and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450.
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014.
- Zhang, J.; Huang, K.; O’Neill, K.L.; Pang, X.; Luo, X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016, 7, e2266.
- Hao, Z.; Duncan, G.S.; Su, Y.-W.; Li, W.Y.; Silvester, J.; Hong, C.; You, H.; Brenner, D.; Gorrini, C.; Haight, J.; et al. The E3 ubiquitin ligase Mule acts through the ATM–p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 2012, 209, 173–186.
- Qi, C.-F.; Kim, Y.-S.; Xiang, S.; Abdullaev, Z.; Torrey, T.A.; Janz, S.; Kovalchuk, A.L.; Sun, J.; Chen, D.; Cho, W.C.; et al. Characterization of ARF-BP1/HUWE1 Interactions with CTCF, MYC, ARF and p53 in MYC-Driven B Cell Neoplasms. Int. J. Mol. Sci. 2012, 13, 6204–6219.
- Ma, W.; Zhao, P.; Zang, L.; Zhang, K.; Liao, H.; Hu, Z. Tumour suppressive function of HUWE1 in thyroid cancer. J. Biosci. 2016, 41, 395–405.
- Zhang, J.; Kan, S.; Huang, B.; Hao, Z.; Mak, T.W.; Zhong, Q. Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 2011, 25, 2610–2618.
- Germain, M.; Milburn, J.; Duronio, V. MCL-1 Inhibits BAX in the Absence of MCL-1/BAX Interaction. J. Biol. Chem. 2008, 283, 6384–6392.
- Warr, M.R.; Acoca, S.; Liu, Z.; Germain, M.; Watson, M.; Blanchette, M.; Wing, S.S.; Shore, G.C. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett. 2005, 579, 5603–5608.
- Myant, K.B.; Cammareri, P.; Hodder, M.C.; Wills, J.; Von Kriegsheim, A.; Győrffy, B.; Rashid, M.; Polo, S.; Maspero, E.; Vaughan, L.; et al. HUWE 1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Mol. Med. 2016, 9, 181–197.
- Pervin, S.; Tran, A.; Tran, L.; Urman, R.; Braga, M.; Chaudhuri, G.; Singh, R. Reduced association of anti-apoptotic protein Mcl-1 with E3 ligase Mule increases the stability of Mcl-1 in breast cancer cells. Br. J. Cancer 2011, 105, 428–437.
- He, G.-Q.; Xu, W.-M.; Liao, H.-J.; Jiang, C.; Li, C.-Q.; Zhang, W. Silencing Huwe1 reduces apoptosis of cortical neurons exposed to oxygen-glucose deprivation and reperfusion. Neural Regen. Res. 2019, 14, 1977–1985.
- Liao, M.; Zhao, J.; Wang, T.; Duan, J.; Zhang, Y.; Deng, X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol. Cancer 2011, 10, 44.
- Stewart, D.P.; Koss, B.; Bathina, M.; Perciavalle, R.M.; Bisanz, K.; Opferman, J.T. Ubiquitin-Independent Degradation of Antiapoptotic MCL-1. Mol. Cell. Biol. 2010, 30, 3099–3110.
- Ding, Q.; He, X.; Hsu, J.-M.; Xia, W.; Chen, C.-T.; Li, L.-Y.; Lee, D.-F.; Liu, J.-C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by β-TrCP Mediates Glycogen Synthase Kinase 3-Induced Tumor Suppression and Chemosensitization. Mol. Cell. Biol. 2006, 27, 4006–4017.
- Senichkin, V.V.; Kopeina, G.S.; Prokhorova, E.A.; Zamaraev, A.V.; Lavrik, I.N.; Zhivotovsky, B. Modulation of Mcl-1 transcription by serum deprivation sensitizes cancer cells to cisplatin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 557–566.
- Yu, X.; Lijun, L.; Xia, Z.; Xie, L.; Ma, X.; Liang, Q.; Liu, L.; Wang, J.; Zhou, X.; Yang, Y.; et al. Targeting MCL-1 sensitizes human esophageal squamous cell carcinoma cells to cisplatin-induced apoptosis. BMC Cancer 2017, 17, 1–13.
- Fletcher, S. MCL-1 inhibitors—Where are we now (2019)? Expert Opin. Ther. Pat. 2019, 29, 909–919.
- Perego, P.; Giarola, M.; Righetti, S.C.; Supino, R.; Caserini, C.; Delia, D.; Pierotti, M.A.; Miyashita, T.; Reed, J.C.; Zunino, F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562.
- Di Pietro, A.; Koster, R.; Eck, W.B.-V.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; De Vries, E.G.; De Jong, S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012, 11, 4552–4562.
- Itahana, Y.; Ke, H.; Zhang, Y. p53 Oligomerization Is Essential for Its C-terminal Lysine Acetylation. J. Biol. Chem. 2009, 284, 5158–5164.
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288.
- Grabbe, C.; Husnjak, K.; Dikic, I. Europe PMC Funders Group. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2013, 12, 295–307.
- Deben, C.; Wouters, A.; De Beeck, K.O.; Bossche, J.V.D.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor Nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679.
- Zanjirband, M.; Edmondson, R.J.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134.
- Zilfou, J.T.; Lowe, S.W. Tumor Suppressive Functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883.
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2015, 7, 4695–4711.
- Alzoubi, S.; Brody, L.; Rahman, S.; Mahul-Mellier, A.-L.; Mercado, N.; Ito, K.; El-Bahrawy, M.; Silver, A.; Boobis, A.; Bell, J.D.; et al. Synergy between histone deacetylase inhibitors and DNA-damaging agents is mediated by histone deacetylase 2 in colorectal cancer. Oncotarget 2016, 7, 44505–44521.
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2016, 36, 1804–1815.
- Ji, P.; Yeh, V.; Ramirez, T.; Murata-Hori, M.; Lodish, H.F. Histone deacetylase 2 is required for chromatin condensation and subsequent enucleation of cultured mouse fetal erythroblasts. Haematologica 2010, 95, 2013–2021.
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622.
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92.
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052.
More
Information
Subjects:
Cell & Tissue Engineering
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
19 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No