2. HUWE1 Modulates the Intrinsic Apoptotic Pathway
A severely threatened genome shifts the DDR from a reparative state towards controlled cell death
[15][9]. Apoptotic signaling follows interconnected patterns that ultimately converge on caspase-executioners for cellular decay. Genotoxins including platins rely on the intrinsic, mitochondrial, pathway to induce cell death. Intrinsic apoptosis is regulated by a delicate balance between pro- and anti-apoptotic members of the Bcl-2 family. These control the release of cytochrome c (cyt c) into the cytoplasm
[16][17][99,100]. In response to genotoxic stress, pro-apoptotic Bcl-2 induction destabilizes the mitochondrial membrane, thereby facilitating the cytoplasmatic release of cyt c and caspase activation
[17][100]. The tumor suppressor p53 is a principal pro-apoptotic non-Bcl-2 protein and DDR effector that promotes this process
[18][101]. Post-translational activation of p53 in response to cellular stress triggers an apoptotic cascade
[19][102].
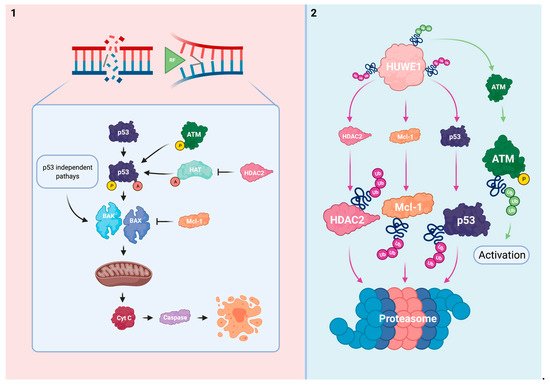
The intrinsic apoptotic pathway is influenced by the effects of HUWE1. Considering this process, HUWE1 directly regulates p53 and Mcl-1, an anti-apoptotic member of the Bcl-2 family. In addition, HUWE1 impairs the post-translational activation of p53 via regulation of ATM and histone deacetylase 2 (HDAC2) (). HUWE1-mediated effects on the cellular ability to induce apoptosis in response to platins might be considered as a potent driver of therapy sensitivity.
Figure 14. HUWE1 influences the intrinsic apoptotic pathway. (1) In response to irreparable DNA damage, p53 is activated via ATM-mediated phosphorylation and HAT-mediated acetylation. HDAC2 inhibits acetylation of p53. The pro-apoptotic BAK/BAX are stimulated by p53 activation and via p53 independent mechanisms. This subsequently results in mitochondrial pore formation and the cytoplasmatic release of cyt c to effectuate apoptosis. The anti-apoptotic Mcl-1 inhibits BAK/BAX. (2) HUWE1 negatively regulates p53, Mcl-1, and HDAC2 via polyubiquitination. HUWE1 positively regulates ATM phosphorylation via polyubiquitination. HUWE1 depletion could result in cellular immortalization and resistance to platins via target overexpression and subsequent ineffective activation of the intrinsic apoptotic pathway.
P53 loss of function occurs frequently in various cancers and can lead to cellular immortalization
[20][103]. Normally, cells maintain p53 at low levels to maintain their proliferative capabilities via mechanisms such as the mouse double minute 2 homolog (MDM2), an E3 Ub ligase. However, in response to cellular stress, p53 is rapidly phosphorylated and stabilized to block MDM2-mediated degradation
[21][104]. In addition to phosphorylation, histone acetyl-transferase (HAT)-mediated p53 acetylation is indispensable for its activation
[22][105]. Activated p53 stimulates multiple proteins including the Bcl-2 homologous antagonist/killer (BAK) and Bcl-2 associated X (BAX) to shift cellular balance towards a pro-apoptotic state
[23][24][106,107]. However, BAK and BAX can also be activated in p53 deficient conditions due to Mcl-1 neutralization, thereby indicating the relevance of the interplay between HUWE1 and intrinsic apoptosis even in a p53 mutant tumor
[25][108].
HUWE1 has been found to directly target p53 for proteasomal degradation in multiple models
[10][12][26][27][28,30,82,109]. This reciprocal relationship at steady state seems to promote the cell’s proliferative capabilities similarly to the MDM2/p53 complex. Conversely, a positive relationship between HUWE1 and p53 has been identified in multiple thyroid cancer models. HUWE1 overexpression increased p53 stability by MDM2 downregulation in thyroid cancer (WRO) cells and mouse xenografts. Ectopic HUWE1 expression in HUWE1 KD thyroid cancer cells sensitized this model to cisplatin and other genotoxins
[28][110].
ATM activation is mediated by HUWE1 in B-cells and MEFs as previously discussed. In HUWE1 deficient and p53 sufficient conditions, these cells failed to successfully trigger a p53 response cascade upon exposure to doxorubicin, etoposide, and y-irradiation due to insufficient levels of activated ATM. Interestingly, the response to dexamethasone, a p53-independent inducer of apoptosis, was not influenced by HUWE1 status in B-cells
[26][82].
HDAC2 has been identified as a HUWE1 target for proteasomal degradation in MEFs
[29][111]. HUWE1 deficiency impaired the ability to induce apoptosis in response to cisplatin or nutlin-3 treatment via HDAC2 enhancement in these cells. Whereas phosphorylated and acetylated p53 accumulated in HUWE1 WT-expressing controls, HUWE1 KO MEFs failed to effectuate these post-translational modifications upon genotoxic stress. Subsequently, these cells could not effectively initiate a downstream p53 response cascade. Reduction of HDAC2 to near WT levels was sufficient to normalize the stress-induced p53 cascade and re-sensitized HUWE1 KO MEFs to cisplatin and nutlin-3 treatment
[29][111]. Taken together, these data imply that loss of HUWE1 impairs the ability to effectively trigger and effectuate p53-mediated apoptosis in response to platins and other genotoxins.
Mcl-1 exerts its anti-apoptotic function primarily by complex formation with the pro-apoptotic BAK and BAX proteins via their shared BH3 domains. Within these complexes BAK and BAX’s ability to destabilize the mitochondrial membrane is impaired
[30][112].
HUWE1′s BH3 domain mostly resembles BAK’s BH3 domain allowing it to act as a dose-dependent regulator of Mcl-1 in HeLa cells
[9][31][27,113]. Whereas the BH3 domain serves as a Mcl-1 docking station, the consecutive action of HUWE1′s catalytic HECT domain ubiquitinates Mcl-1 for proteasomal degradation
[9][27]. Cellular Mcl-1 has been reported to accumulate in HUWE1 deficient conditions
[9][29][31][32][33][27,111,113,114,115]. Interaction between HUWE1 and other Bcl-2 family members was not observed in HeLa cells
[9][31][27,113]. However, HUWE1 KD in an ischemic cortical neuron model modulated Bcl-2 and BAX additionally to Mcl-1 which further promoted the anti-apoptotic phenotype
[34][116]. HUWE1 might thus have a more comprehensive role in regulation of Bcl-2 members.
The direct relationship between HUWE1, Mcl-1, and the response to platins has been described in several reports
[9][29][32][35][27,111,114,117]. HUWE1 KD in HeLa cells impaired the apoptotic response upon UV-irradiation, etoposide, or cisplatin exposure via Mcl-1 enhancement. Of the investigated genotoxic agents, cisplatin proved to be the least potent to trigger apoptosis
[9][27]. In line with these findings, Mcl-1 reduction to WT levels re-sensitized HUWE1 KO MEFs to cisplatin treatment
[29][111]. Others indicated that bile salt-induced Mcl-1 phosphorylation enhanced Mcl-1 stability by blocking HUWE1-mediated degradation in human liver cancer (HepG2) cells. The subsequent inability to effectuate apoptosis upon cisplatin treatment could be rescued by Mcl-1 KD in these cells
[35][117]. Similarly to the observations in cell models, intestinal crypts of transgenic colorectal cancer HUWE1 KO mice displayed a decreased cisplatin sensitivity by Mcl-1 upregulation
[32][114]. HUWE1 deficiency thus protects cells from undergoing apoptosis in response to platins and other genotoxins via Mcl-1 upregulation.
Mcl-1 is considered one of HUWE1′s principal targets in oncology. Next to HUWE1, Mcl-1 turnover is also mediated by the β-transducin repeat-containing E3 Ub protein ligase and Ub-independent pathways
[36][37][118,119]. In addition to the direct evidence between the HUWE1/Mcl-1 axis and cisplatin sensitivity, Mcl-1 overexpression is a well-known inducer of resistance to platins and other genotoxins
[38][39][120,121]. The potential anti-neoplastic use of Mcl-1 inhibitors is currently being investigated
[40][122].
Non-functional mutagenic p53 can impair the apoptotic response and is linked to platinum-based therapy resistance
[20][41][103,123]. However, the exact role of p53 in modulating sensitivity to platins is complex and cell context-dependent
[42][124]. Mutagenic loss of p53 function is accompanied by conformational changes that influence its susceptibility to post-translational modifications for protein activation. Interestingly, the impaired ability to acetylate mutated p53 is linked to its decreased functionality
[43][125]. In addition, overexpression and cytoplasmatic mislocalization of p53 have been linked to platinum-based therapy resistance by inhibiting caspase effectors
[44][126]. Similarly to mutagenic p53, oncogenic loss of HUWE1 might decrease the cellular responsiveness to platins via impaired p53 activation. However, the reported relationship between HUWE1 and p53 varies among cell types and depends on the interplay with MDM2
[10][12][26][27][28][28,30,82,109,110]. This strengthens the concept that Ub network organization varies in spatiotemporal dimensions
[45][127]. Whereas inhibition of MDM2 with nutlin-3 sensitizes cells to platins, this mechanism of action relies on enhancing p53 levels rather than promoting its post-translational activation
[46][47][128,129]. Nonetheless, it highlights the potential of targeting UPS mediators to promote p53-mediated apoptosis. Complementary to its principal role in apoptotic signaling, one should note that p53 has a broad spectrum of other effects within the DDR
[48][130].
Next to the direct evidence on HDAC2 as a cisplatin desensitizer upon loss of HUWE1, HDAC2-induced resistance to cisplatin and other DNA damaging agents is supported within other conditions
[49][50][131,132]. Other HDACs are however involved in the post-translational modification of p53 which further indicates the potential existence of cellular rescue and compensation mechanisms
[51][133]. In addition to its non-histone effects on p53, HDAC2-mediated deacetylation of histones results in DNA condensation
[52][134]. This impairs the DNA accessibility for platins to induce DNA damage
[53][135]. The use of HDAC inhibitors is an emerging field of cancer drug research that could be especially promising when applied synergistically with established cancer drugs such as platins
[54][55][136,137].
Platinum’s primary mode of action is inseparably connected to the intrinsic apoptotic pathway. Altogether, the direct and indirect regulatory effects of HUWE1 on Mcl-1 and p53 propose an important role for HUWE1 in successfully triggering intrinsic apoptosis in response to platins. This suggests that HUWE1 has the potential to act as a biomarker to assess an individual’s response to platins. Moreover, stimulation of HUWE1 could induce the intrinsic apoptotic pathway which further promotes HUWE1 as a sensitizer of platinum-based therapy.