Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alfredo Cappariello | + 1913 word(s) | 1913 | 2021-07-15 10:40:30 | | | |
| 2 | Amina Yu | Meta information modification | 1913 | 2021-07-19 09:39:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cappariello, A. High-Fat Diet and Small Intestine. Encyclopedia. Available online: https://encyclopedia.pub/entry/12163 (accessed on 07 February 2026).
Cappariello A. High-Fat Diet and Small Intestine. Encyclopedia. Available at: https://encyclopedia.pub/entry/12163. Accessed February 07, 2026.
Cappariello, Alfredo. "High-Fat Diet and Small Intestine" Encyclopedia, https://encyclopedia.pub/entry/12163 (accessed February 07, 2026).
Cappariello, A. (2021, July 19). High-Fat Diet and Small Intestine. In Encyclopedia. https://encyclopedia.pub/entry/12163
Cappariello, Alfredo. "High-Fat Diet and Small Intestine." Encyclopedia. Web. 19 July, 2021.
Copy Citation
The high-fat diet (HFD) of western countries has dramatic effect on the health of several organs, including the digestive tract, leading to the accumulation of fats that can also trigger a chronic inflammatory process, such as that which occurs in non-alcohol steatohepatitis. The effects of a HFD on the small intestine, the organ involved in the absorption of this class of nutrients, are still poorly investigated.
intestine
diet
lipids
lipid droplets
fat
intestinal inflammation
small bowel
1. Introduction
The small intestine is actively involved in the metabolism of dietary fats, such as cholesteryl esters (CEs), phospholipids (PLs), and triglycerides (TGs); among these, approximately 90% of the total fat calories are provided by TGs [1][2][3][4]. The first step in lipid digestion and emulsification occurs in the mouth and stomach due to the combined action of salivary and gastric lipases. Pancreatic lipases and bile acids then cooperate to further digest and emulsify lipids, facilitating their absorption in the small intestine. Lipases split TGs into fatty acids and monoglycerides, which are packaged in mixed micelles, making lipids absorbable by the intestinal epithelium [1][2][3][4]. Once inside the small intestine, they are resynthesized, and fatty acids and monoglycerides reach the endoplasmic reticulum membrane, where they have two fates: a portion of them get into the endoplasmic reticulum and are transported into the Golgi apparatus and secreted into the bloodstream as chylomicrons [4][5][6][7]. The other portion of lipids are stored in cytosolic lipid droplets (CLDs), which are spherical particles surrounded by a phospholipid monolayer and covered on the surface by several proteins, mainly members of the perilipin family. In physiological conditions, CLDs are temporarily accumulated in the cytosol of enterocytes as an energy reservoir and are released for energy supply during the intestinal fasting state [4][5][6][7]. However, when excessive accumulation of lipids occurs, as the result of an unbalanced diet or alterations in lipogenesis/lipolysis processes, severe diseases may result [8][9][10][11][12][13][14][15][16][17][18].
A surplus of TGs induces liver injury, such as nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), and even cirrhosis and hepatocarcinoma [12][16][17][18]. They may also be responsible for pancreatic damage by reducing apoptosis of β-cells, with the consequent reduction in insulin secretion [14][15]. Moreover, the accumulation of TGs in skeletal muscle leads to insulin resistance and a reduction of glucose uptake [10][19], while accumulation in the myocardium results in cardiomyopathy [9][11].
Although the intestinal tract is known to be directly involved in lipid metabolism, the mucosal alterations induced by an enriched fat diet are still limited [20][21].
Indeed, several studies have focused attention on the changes in intestinal microbiota and the exacerbation of several intestinal disorders (i.e., food allergies, inflammatory bowel disease (IBD), or colorectal cancer) [22][23][24][25], while a few studies have evaluated the impact of a high-fat diet (HFD) on intestinal inflammation [26][27][28][29].
Lipid metabolism is regulated by several molecules, including peroxisome proliferator-activated receptor-γ (PPAR-γ), a master gene of adipogenesis and adipocyte differentiation that is also involved in the regulation of many processes, such as cell proliferation, differentiation, inflammation, and fibrosis [30][31][32]; for this reason, PPAR-γ could be a promising therapeutic target. It has been demonstrated that rosiglitazone and other molecules that are able to enhance PPAR-γ functions lead to an improvement in inflammation and metabolic alterations induced by excessive fat intake [30][31][32].
A HFD also increases the expression of adipokines involved in lipid metabolism, such as leptin, which are responsible for a downregulation of PPAR-γ and the upregulation of proinflammatory cytokines like TNF-α and IL-6 through the PI3K/mTOR pathway [33][34][35][36].
In previous studies, we demonstrated that the abuse of an 18-month long chronic hyperlipidemic diet induced severe metabolic dysfunctions altering the mice’s body weight, liver weight/body weight ratio, serum parameters like ALT, AST, triglycerides, and cholesterol and prompted the liver parenchyma towards nonalcoholic steatohepatitis (NASH) and hepatocarcinoma [17][18]. On this basis, we hypothesized that the same dietary regimen may directly induce also alterations of bowel wall morphology and that the accumulation of fat in the mucosa may induce mucosal damage and may trigger the onset of intestinal inflammation. This study aimed to make a pairwise evaluation of the impact of the chronic administration of an HFD w/Suc on the small intestine of the same mice already evaluated for liver phenotype in our previous studies [17][18].
2. Morphological Changes in the Small Intestine Architecture Induced by a High Fat with Sucrose Diet
Histological analyses by H&E staining showed a marked accumulation of lipid droplets in the mucosa of HFD w/Suc-fed mice, leading to a disruption of normal intestinal architecture (Figure 1A).
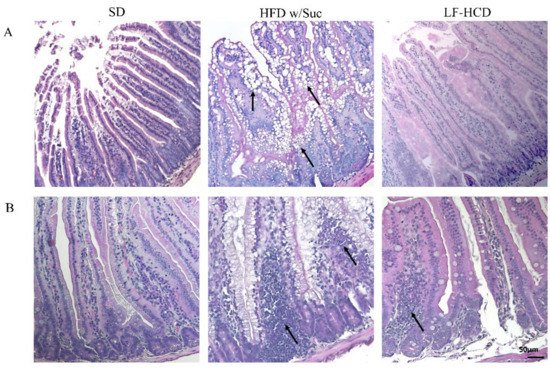
Figure 1. Hematoxylin and Eosin staining of proximal tract of the small intestine. Original magnification: 20×; scale bar: 50 μm. (A) A marked accumulation of lipid droplets was present in the intestinal mucosa of HFD w/Suc -fed mice (arrows) compared to SD-fed and LF-HCD-fed mice. (B) A massive infiltration of inflammatory cells was found in HFD w/Suc-fed mice compared to the SD group, while LF-HCD-fed mice showed only scanty and mild inflammatory foci (arrows). These images are representative of n = 5 SD, n = 10 HFD w/Suc, n = 10 LF-HCD mice.
Additionally, in the HFD w/Suc mice, a spread inflammatory infiltrate was found across the mucosa (Figure 1B), whereas no signs of CLD deposition and relevant inflammatory foci were detected in the SD group (Figure 1A,B). Interestingly, the analysis of the small intestine from mice fed with a hypercaloric low-fat (4.8%), high-sucrose (61.12%) diet did not show significant signs of tissue alterations and lipidic accumulation or signs of significant inflammation. These findings suggest that the lipidic content of the diet can specifically induce intestinal alterations.
To highlight the dietary effects on intestinal epithelial barrier components, PAS staining elective for goblet cells was also undertaken (Figure 2A). The goblet cells secrete numerous mucins that form a thin layer of mucus adhering to the mucosal surface, which represents an important component of the intestinal mucosal barrier. The quantitative evaluation revealed a significant reduction of goblet cells in HFD w/Suc mice compared to SD mice (Figure 2B).
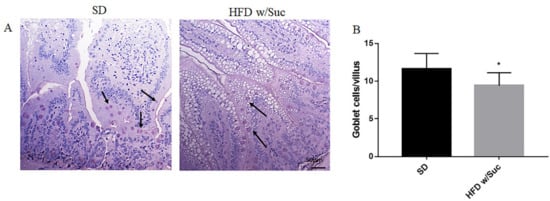
Figure 2. (A) PAS staining of proximal tract of the small intestine. Original magnification: 20×; scale bar: 50 μm. (B) Quantification of goblet cells. HFD w/Suc mice showed a reduction of goblet cell numbers compared to the SD group (arrows). * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
Fibrosis was absent in both the SD and HFD w/Suc mice (Figure 3).
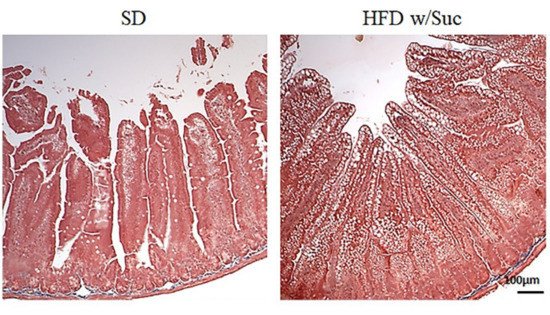
Figure 3. Masson’s trichrome staining of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). No signs of fibrosis were found in either the SD or the HFD w/Suc -fed mice. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
3. Evaluation of Protein Changes Induced by a High-Fat with Sucrose Diet
To confirm the presence of lipids, we performed immunohistochemistry analysis for perilipin 1, which is one of the CDL surface proteins (Figure 4A).
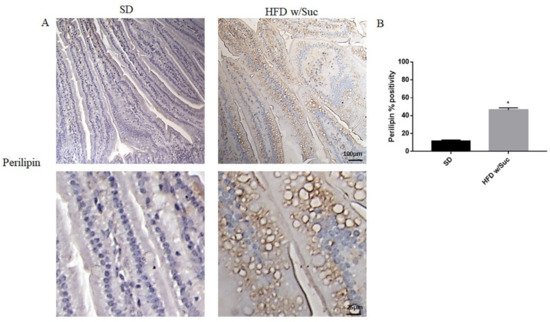
Figure 4. (A) Immunohistochemistry for perilipin 1 of proximal tract of the small intestine. (Upper: original magnification: 10×; scale bar: 100 μm, bottom: magnification 40×; scale bar: 25 μm). (B) The expression of perilipin 1 was significantly increased in HFD w/Suc mice compared to the SD group. * p <0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
HFD w/Suc mice showed increased perilipin expression compared to the control group, mainly localized on the rim of the lipid droplets present in the intestinal mucosa layer (Figure 4A). The increase of perilipin in the HDF w/Suc group was confirmed by semi-quantitative analysis (Figure 4B).
To confirm that a HFD w/Suc leads to an overexpression of leptin, we assessed this molecule and its receptor (Figure 5).
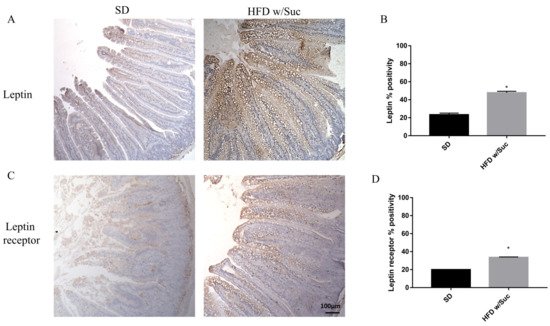
Figure 5. (A,C) Immunohistochemistry for leptin and leptin receptor of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). Immunostaining of both leptin and its receptor showed an increase in HFD w/Suc mice compared to the SD group. (B,D) Semiquantitative analyses of leptin and leptin receptors confirmed the increased expression of these two molecules. * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
Immunostaining of leptin is noticeably increased in HFD w/Suc-fed mice compared to the SD group (Figure 5A,B). Evaluation of leptin receptor also showed significant differences between SD and HFD w/Suc-fed mice (Figure 5C,D).
Furthermore, we performed an immunohistochemistry evaluation of adiponectin, a molecule that generally counterbalances the activity of leptin (Figure 6A).
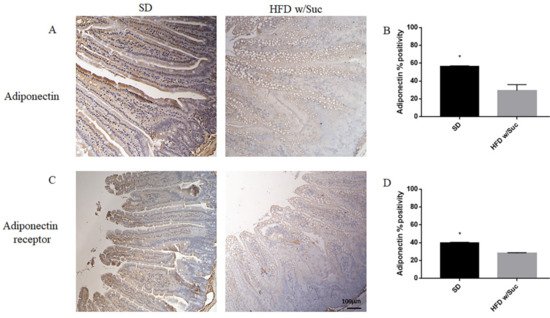
Figure 6. (A,C) Immunohistochemistry of adiponectin and adiponectin receptor of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). (B,D) The expression of adiponectin and its receptor was significantly reduced in HFD w/Suc mice compared to the SD group. * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
The analysis revealed a significant reduction of adiponectin expression in the HFD w/Suc group compared to control mice (Figure 6A), as validated by the semiquantitative analysis (Figure 6B).
Similarly, adiponectin receptor immunopositivity was reduced in the HFD w/Suc group compared to the SD mice (Figure 6C,D).
Immunohistochemistry of PPAR-γ showed a reduction of positivity in HFD w/Suc -fed mice compared to the SD group (Figure 7A), as confirmed by the semiquantitative analysis (Figure 7B). These data are in line with the anti-inflammatory role of this transcriptional factor [32].
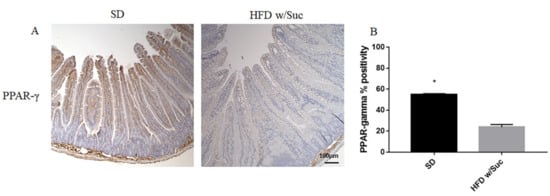
Figure 7. (A) Immunohistochemistry of PPAR-γ of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). (B) PPAR-γ was significantly reduced in HFD w/Suc mice compared to the SD group. * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
Since the increase of lipid droplets was associated with a concomitant increase of mucosal inflammatory infiltrate in HDF w/Suc mice, we performed immunohistochemistry analysis for the proinflammatory mediators PI3K, phosphorylated-mTOR (p-mTOR), and phosphorylated Akt (p-Akt) (Figure 8) [37][38][39].
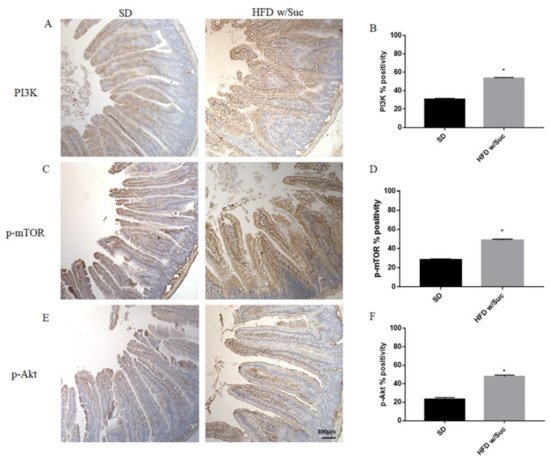
Figure 8. (A,C,E) Immunohistochemistry of PI3K, p-mTOR, and p-Akt of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). (B,D,F) All the tested molecules were increased in the HFD w/Suc group compared to SD mice. * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
Immunostaining of PI3K, p-mTOR, and p-Akt was increased in the HFD w/Suc group compared to the SD-fed mice (Figure 8A,C,E), which was supported by the semiquantitative analyses of each of these examined molecules (Figure 8B,D,F). In addition, we evaluated the expression of TNF-α, one of the main proinflammatory factors (Figure 9).
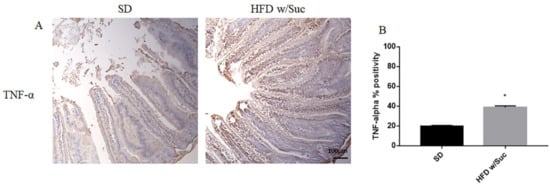
Figure 9. (A) Immunohistochemistry of TNF-α of proximal tract of the small intestine. (Original magnification: 10×; scale bar: 100 μm). (B) TNF-α showed an increase in the HFD w/Suc group compared to SD mice. * p < 0.05. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
Immunohistochemistry (Figure 9A) and semiquantitative evaluation (Figure 9B) revealed a significant increase of TNF-α in HFD w/Suc-fed mice compared to the SD group, confirming the onset of an inflammatory process after the consumption of a hyperlipidic diet.
Finally, to investigate the integrity of the intestinal epithelium, we evaluated the expression of Zonulin-1, a key molecule in the formation and remodeling of tight junctions and the maintenance of the intestinal barrier. Immunohistochemical analysis showed a marked decrease in the expression of zonulin-1 in the epithelium of the group of mice receiving HFD w/Suc compared to control mice (Figure 10), supporting the hypothesis that the hyperlipidic diet may alter the functions of the intestinal epithelial barrier.
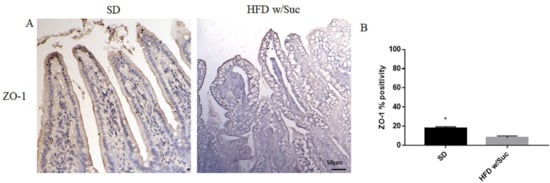
Figure 10. (A) Immunohistochemistry for Zonulin 1 (ZO-1) of proximal tract of the small intestine. (Original magnification: 20×; scale bar: 50 μm). (B) ZO-1 showed an increase in the SD group compared to HFD w/Suc mice. These images are representative of n = 5 SD, n = 10 HFD w/Suc mice.
References
- Phan, C.T.; Tso, P. Intestinal lipid absorption and transport. Front. Biosci 2001, 6, D299–D319.
- Mu, H.; Hoy, C.E. The digestion of dietary triacylglycerols. Prog. Lipid Res. 2004, 43, 105–133.
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194.
- Hussain, M.M. Intestinal Lipid Absorption and Lipoprotein Formation. Curr. Opin. Lipidol. 2014, 25, 200–206.
- Ko, C.V.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183.
- Demignot, S.; Beilstein, F.; Morel, E. Triglyceride-rich lipoproteins and cytosolic lipid droplets in enterocytes: Key players in intestinal physiology and metabolic disorders. Biochimie 2014, 96, 48–55.
- Beilstein, F.; Carrière, V.; Leturque, A.; Demignot, S. Characteristics and functions of lipid droplets and associated proteins in enterocytes. Exp. Cell Res. 2016, 340, 172–179.
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 2001, 50, 1612–1617.
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514.
- Gaborit, B.; Kober, F.; Jacquier, A.; Moro, P.J.; Cuisset, T.; Boullu, S.; Dadoun, F.; Alessi, M.C.; Morange, P.; Clement, K.; et al. Assessment of epicardial fat volume and myocardial triglyceride content in severely obese subjects: Relationship to metabolic profile, cardiac function and visceral fat. Int. J. Obes. 2012, 36, 422–430.
- Gastaldelli, A.; Morales, M.A.; Marraccini, P.; Sicari, R. The role of cardiac fat in insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 523–528.
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560.
- Morelli, M.; Gaggini, M.; Daniele, G.; Marraccini, P.; Sicari, R.; Gastaldelli, A. Ectopic fat: The true culprit linking obesity and cardiovascular disease? Thromb. Haemost. 2013, 110, 651–660.
- Gastaldelli, A. Visceral adipose tissue and ectopic fat deposition. In Handbook of Obesity; Bray, G.A., Bouchard, C., Eds.; CRC Press: Boca Raton, FL, USA, 2014; Volume 1, pp. 237–248.
- Yki-Jarvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910.
- Gaggini, M.; Saponaro, C.; Gastaldelli, A. Not all fats are created equal: Adipose vs. Ectopic fat, implication in cardiometabolic diseases. Horm. Mol. Biol. Clin. Investig. 2015, 22, 7–18.
- Tessitore, A.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Pompili, S.; Cicciarelli, G.; Barnabei, R.; Capece, D.; Zazzeroni, F.; Capalbo, C.; et al. Development of hepatocellular cancer induced by long term low fat-high carbohydrate diet in a NAFLD/NASH mouse model. Oncotarget 2017, 8, 53482–53494.
- Pompili, S.; Vetuschi, A.; Gaudio, E.; Tessitore, A.; Capelli, R.; Alesse, E.; Latella, G.; Sferra, R.; Onori, P. Long-term abuse of a high-carbohydrate diet is as harmful as a high-fat diet for development and progression of liver injury in a mouse model of NAFLD/NASH. Nutrition 2020, 75–76, 110782.
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474.
- Ahmad, R.; Rah, B.; Bastola, D.; Dhawan, P.; Singh, A.B. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci. Rep. 2017, 7, 5125.
- Tuganbaev, T.; Mor, U.; Bashiardes, S.; Liwinski, T.; Nobs, S.P.; Leshem, A.; Dori-Bachash, M.; Thaiss, C.A.; Pinker, E.Y.; Ratiner, K.; et al. Diet Diurnally Regulates Small Intestinal Microbiome-Epithelial-Immune Homeostasis and Enteritis. Cell 2020, 182, 1441–1459.
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of High-Fat-Diet on Gut Microbiota: A Driving Force for Chronic Disease Risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520.
- Araújo, J.R.; Tomas, J.; Brenner, C.; Sansonetti, P.J. Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie 2017, 141, 97–106.
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91.
- Tanaka, S.; Nemoto, Y.; Takei, Y.; Morikawa, R.; Oshima, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; Stutte, S.; et al. High-fat diet-derived free fatty acids impair the intestinal immune system and increase sensitivity to intestinal epithelial damage. Biochem. Biophys. Res. Commun. 2020, 522, 971–977.
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649.
- McAlester, A.H.; Kim, M.; Song, R.H.; Wu, W.; Diehl, G. Acute high fat diet disrupts intestinal barrier repair. J. Immunol. 2018, 200, 53.10.
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G453–G462.
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426.
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566.
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.B.; Ducroc, R.; Regnault, B.; Kun, C.; Duszka, T.K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-γ pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943.
- Vetuschi, A.; Pompili, S.; Gaudio, E.; Latella, G.; Sferra, R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8839–8848.
- Hara, K.; Kubota, N.; Tobe, K.; Terauchi, Y.; Miki, H.; Komeda, K.; Tamemoto, H.; Yamauchi, T.; Hagura, R.; Ito, C.; et al. The role of PPARg as a thrifty gene both in mice and humans. Br. J. Nutr. 2000, 84, S235–S239.
- Cabrero, A.; Cubero, M.; Llaverías, G.; Alegret, M.; Sánchez, R.; Laguna, J.C.; Vázquez-Carrera, M. Leptin down-regulates peroxisome proliferator-activated receptor gamma (PPAR-gamma) mRNA levels in primary human monocyte-derived macrophages. Mol. Cell Biochem. 2005, 275, 173–179.
- Iikuni, N.; Lam, Q.L.K.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79.
- Narayana, P.B.; Fazolini, N.P.B.; Cruz, A.L.S.; Werneck, N.B.F.; Viola, J.P.B.; Maya-Monteiro, C.M.; Bozza, P.T. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015, 16, 2667–2676.
- Tokuhira, N.; Kitagishi, Y.; Suzuki, M.; Minami, A.; Nakanishi, A.; Ono, Y.; Kobayashi, K.; Matsuda, S.; Ogura, Y. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy (Review). Int. J. Mol. Med. 2015, 35, 10–16.
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409.
- Radwan, R.R.; Karam, H.M. Resveratrol attenuates intestinal injury in irradiated rats via PI3K/Akt/mTOR signaling pathway. Environ. Toxicol. 2020, 35, 223–230.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
19 Jul 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No