 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandros Makis | + 3085 word(s) | 3085 | 2021-06-24 07:35:13 | | | |
| 2 | Dean Liu | Meta information modification | 3085 | 2021-07-13 03:07:31 | | |
Video Upload Options
Beta-thalassemia (β-thalassemia) is an autosomal recessive inherited disease characterized by decreased production of the β-globin chains of hemoglobin (Hb) A. The normal structure of HbA is two α- and two β-globin chains. Individuals with β-thalassemia are either homozygous or double heterozygotes for mutations in the β-globin gene.
1. Introduction
Beta-thalassemia (β-thalassemia) is an autosomal recessive inherited disease characterized by decreased production of the β-globin chains of hemoglobin Individuals with β-thalassemia are either homozygous or double heterozygotes for mutations in the β-globin gene. The severity of the disease depends on the type of mutation in the β-globin gene and the extent of impairment of β-globin chain production. β-thalassemia can be classified as transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT) according to the severity of anemia and the need for transfusions.
These iron-containing insoluble bodies induce the generation of reactive oxygen species that are deleterious to the cell membrane structures of erythroid cells. The oxidative stress leads to the premature apoptosis of the erythroblasts, which is called ineffective erythropoiesis [1][2]. The changes in membrane proteins of mature red blood cells (RBC), especially the increased expression of phosphatidylserine, lead to hemolysis from the macrophages of the reticuloendothelial system. Furthermore, the overactivation of transforming growth factor-β (TGF-β), which acts as an inhibitor of the final stage of erythropoiesis, has an important role in the process of ineffective erythropoiesis [3].
Ineffective erythropoiesis and peripheral hemolysis lead to severe anemia, tissue hypoxia, and a reactive production of erythropoietin (EPO) with a consequent compensatory increase of the number of bone marrow erythroblasts and extramedullary hematopoiesis with characteristic hepatosplenomegaly. The activated erythroblasts release several proteins, such as erythroferrone (ERFE), which inhibit hepcidin, the master regulator of iron homeostasis. The consequence of low hepcidin levels is the increased intestinal absorption of iron, which, along with chronic hemolysis and RBC transfusions, leads to progressive tissue iron deposition. Excessive free iron has a negative impact in erythropoiesis, creating a vicious cycle between ineffective erythropoiesis and increased iron absorption [4][5][6][7][8][9].
2. Curative Treatments
The Pesaro criteria are applicable to patients below the age of 16 and include three variables related to iron toxicity: regularity of iron chelation, the presence of hepatomegaly, and the presence of liver fibrosis. Allo-HSCT in thalassemic patients with high-risk criteria poses difficulties due to elevated rates of graft rejection and transplant-related mortality [10]. Adults with TDT will always be at high risk, but several new conditioning regimens are being evaluated in clinical trials as an effort to improve the transplant outcomes (NCT01050855, NCT00920972, NCT02435901). Favorable results have been reported using modified or reduced intensity conditioning (e.g., treosulfan/thiotepa/fludarabine) or even nonmyeloablative regimens.
The preferable source of stem cells is the bone marrow rather than peripheral blood, possibly due to lower risk of development of chronic graft-versus-host disease. Peripheral blood stem cells have also been used in an effort to decrease the possibility of graft rejection in high-risk thalassemic patients (NCT02105766). Optimally, fully matched sibling donors are preferred, but matched unrelated donors might also be considered (NCT01049854), as well as related cord blood transplants in patients with low-risk criteria. Unrelated umbilical cord blood cells and haploidentical transplants should ideally be performed in clinical trial setting (NCT02126046, NCT00977691, NCT00408447, NCT02504619).
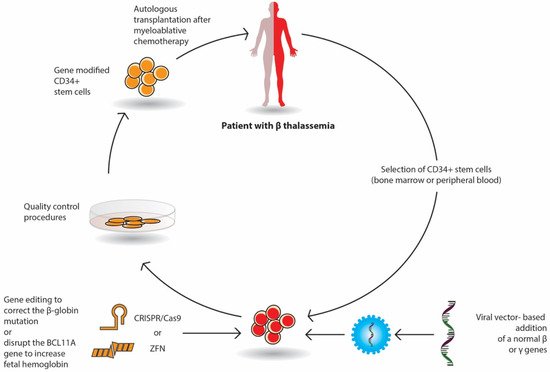
Gene therapy by autologous transplantation of genetically manipulated hematopoietic stem cells is a very promising curative option in β-thalassemia. Among them, lentivirus vectors have been used in thalassemic mouse models and in erythroid stem cells from thalassemic patients. These studies have focused on β or γ-globin addition; the increased expression of γ-globin-activating transcription factors; the silencing of DNA- or RNA-binding proteins that inhibit the expression of γ-globin repressors, such as BCL11A; and the genome editing of β-globin mutations or γ-globin repressors (Figure 1). Currently, several ongoing clinical trials have demonstrated promising results.
The selected CD34+ erythroid progenitor cells (bone marrow or peripheral cells) of the patient are genetically modified either by the viral vector-based addition of a normal β or γ gene or by gene editing with nucleases (Crisp/Cas9 or ZFN), which repair the β globin mutation or delete genomic regions of the BCL11A gene in order to reactivate fetal Hb (HbF) production. The genetically corrected CD34+ stem cells are prepared for autologous transplantation after strict quality control procedures. The patient receives proper myeloablative chemotherapy and then the stem cells are infused in the patient.
In gene addition, a retroviral or lentiviral vector that encloses the regulatory elements and the β- or γ-globin gene area is inserted into previously collected autologous CD34+ erythroid progenitor cells ex vivo and then infused to the patient who has received a myeloablative busulfan conditioning. Following engraftment, it is expected that β-globin or γ-globin production will be restored, and the α/β imbalance will be reduced (Figure 1).
The outcomes of two phase 1/2 clinical trials (HGB-204/NCT01745120 and HGB-205/NCT02151526) using the LentiGlobin BB305 vector in 22 TDT patients (12–35 years of age) were published in 2018 [11]. Regarding the non–β0/β0genotype, all but 1 of the 13 patients became transfusion independent after a median period of 2 years (range, 15–42 months) after the procedure. As expected, the results were different in patients with the most severe genotype (β0/β0or two copies of the IVS1-110 mutation), where transfusion independence requires significantly higher levels of Hb production. In these patients the median transfusion need per year was declined by two thirds, and three patients became transfusion independent.
In order to address this problem, especially in β0/β0patients, the sponsor of the trial, bluebird bio, has significantly improved the protocol with the addition of small molecules to the transduction process. This change has a positive effect on the vector copy number in the ongoing phase 3 trials (HGB 207 and HGB-212). non-β0/β0 patients, leading to transfusion independence with Hb levels > 9 Based on these positive outcomes and satisfactory safety reports, the lentiglobin gene therapy (Zynteglo) received conditional EMA in June 2019 for TDT patients ≥ 12 years of age with a non-β0/β0genotype who are eligible for stem cell transplantation but do not have a matching related donor.
The primary results of the HGB 212 phase 3 trial from 11 patients with either a β0or IVS-I-110 mutation at both alleles of the HBB gene showed that three of four patients stopped transfusions for ≥ 6 months with Hb levels of 10.5–13.6 g/dL at the last visit [12]. After 2 years of follow-up in these four studies (HGB-204, HGB-205, HGB 207, and HGB-212), a long-term follow-up study (LTF-303, NCT02633943) was initiated. The interim results of 32 patients have shown that 64% of the patients from the HGB-204/HGB-205 studies and 90% of the patients of the HGB 207/HGB-212 studies achieved durable and stable transfusion independency [13]. and HGB-212 showed that children achieved transfusion independence with comparable rates and safety as in adults [14].
Another lentiviral gene-insertion phase 1/2 trial (NCT02453477, SR-TIGET) was recently published [15]. Nine patients (three adults, six children) with β0or severe β+mutations received, with intrabone administration, transduced GLOBE lentiviral vector CD34+ cells. Satisfactory hematopoietic engraftment was achieved in all the patients, with no clonal dominance. Three children stopped transfusions, and the adults also exhibited a significant decrease in transfusion requirements.
A different gene insertion approach has been developed at Boston Children’s Hospital. The target is the Bcl11a gene with the use of RNA interference technology. A short hairpin RNA sequence that represses the Bcl11a gene is transduced into CD34+ erythroid progenitor cells using a lentivirus. A relevant clinical trial has been started for sickle cell disease patients [16].
The Bcl11a gene located on chromosome 2 is a promising target for gene editing techniques. Both patients had rapid hematopoietic engraftments with elevated HbF levels [17]. The neutrophil and platelet engraftment occurred at the end of the first month, and all patients stopped receiving transfusions after the second month. The first patient enrolled in the study has remained without the need for transfusion for over 15 months.
3. “Disease-Modifying” (Non-Curative) Treatments
The TGF-β signaling is significant for the regulation of essential cellular pathways, especially in the bone and hematopoietic tissue [18], and comprises four similar protein groups: activins, TGF-β, GDFs (growth and differentiating factors), and BMP (bone morphogenetic proteins). The effect of these proteins on the erythroid lineage can be either inductive (TGF-β, BMP4) Their receptors are serine/threonine kinases that stimulate intracellular paths by recruiting the Sma and Mad related proteins (Smad). TIF1γ stimulates the expression of the key erythroid transcription factor GATA-1 and promotes the differentiation of erythroid lineage [19]
Therefore, the Smad2/3 signaling has appeared as a significant regulatory mechanism of erythropoiesis with a balancing activity according to the implicated pathway. It seems that this orchestrated signaling is necessary for the regular differentiation and cessation of erythropoiesis. The overactivation or dysregulation of Smad2/3-Smad4 signaling has been implicated in illnesses with anemia caused by ineffective erythropoiesis, such as myelodysplastic syndromes or β-thalassemia [3]. Therefore, inhibitors of the TGF-β family that sequester the Smad2/3-Smad4 signaling could promote the end phase of erythropoiesis with a beneficial result in these diseases.
Altered receptors of activin (activin receptor-II trap ligands) have been derived from the colligation of the extracellular part of the activin receptor (ActRIIA or ActRIIB) with the Fc part of IgG immunoglobulin. (sotatercept) is an Fc-IgG fusion trap that derives from the combination of the extracellular fragment of ActRIIA with the Fc part of human IgG. ACE-536 (luspatercept) is a fusion of an altered extracellular domain of ActRIIB with the Fc region of human IgG1, while RAP-536 involves mouse IgG2a [20] RAP-536 fusion proteins have been used in mouse models with thalassemia, with beneficial effects on the reduction of ineffective erythropoiesis, splenomegaly, and iron overload. the promotion of erythroid differentiation [21][22][23][24].
The use of luspatercept in adult thalassemia subjects has been initially evaluated in a phase 2 trial (NCT01749540). In this study, the subcutaneous use of luspatercept improved Hb levels and reduced transfusion requirements [25]. The most common grade 1 to 2 adverse events were headache, bone pain, and myalgia [25]. Comparable results were obtained with sotatercept; however the drug was not selected for further phase 3 studies, mainly because, distinct to luspatercept, it is a less specific activin receptor-II ligand trap, may be less efficacious in treating anemia and have more off-target effects [26].
The trial was designed to define the efficacy and safety of luspatercept in TDT adults [27], who were randomized to obtain subcutaneously either luspatercept at an initial dose of 1.0 mg/kg with a gradual increase up to 1.25 mg/kg or a placebo every 3 weeks for ≥ 48 weeks. The primary endpoint was a ≥ 33% reduction in transfusions, with a reduction of ≥ 2 RBC units during weeks 13 to 24. The secondary endpoints comprised a ≥ 33% reduction in transfusion load and at least 2 RBC units at weeks 37 to 48, and a ≥ 50% reduction in transfusions and at least 2 RBC units at weeks 13 to 24. The primary endpoint was significantly achieved in the luspatercept group: 21.4% vs. 4.5% achieved a reduction of at least 33% in the transfusions during weeks 13 to 24 [27].
While response rates were lower in patients with the most severe disease (β0/β0), clinically significant declines in transfusion load were observed across genotypes. A ≥ 33% decrease in the transfusions at weeks 37–48 was accomplished by 19% of the luspatercept group compared to 3.6% of the placebo group. Regarding the additional target of achieving a ≥ 33% decrease in transfusions throughout any successive 12 weeks of the trial, 70.5% of the luspatercept group reached this level compared to 29% of the placebo group, while for any 24-week period, the difference was 41.1% and 2.7%, respectively. Among patients who achieved a decline of at least 33% during any 12 weeks, 80.4% had at least two episodes of response, and 51.3% had at least four episodes of response.
In order to study the safety and pharmacokinetics in TDT children, a phase 2 clinical trial (NCT04143724) has been planned but is not yet recruiting patients. A major concern in children is the potential toxicity of luspatercept in the developing hematopoietic system and other vital organs. Furthermore, interfering with the BMP pathway may compromise growth in an immature skeletal system.
Luspatercept could also have a place in the treatment of NTDT patients. The BEYOND trial is a phase 2 study to define the effectiveness and safety of luspatercept in adults with NTDT (NCT03342404) [28]. The primary objective is the increase of mean Hb without any transfusions over a 12-week period, from week 13 to 24, compared to the initial phase.
In summary, the subcutaneous use of luspatercept every 3 weeks demonstrated significant reductions in RBC transfusion burden in TDT adult patients and was granted FDA (Food and Drug Administration) and EMA (European Medicines Agency) approval for TDT patients. It seems that luspatercept will have a significant place in the real-life management of TDT patients.
Pyruvate kinase (PK) is the enzyme that plays a significant role in the last stage of glycolysis in the RBC, the conversion of phosphoenolpyruvate to pyruvate in order to generate ATP. In patients with PK deficiency, an autosomal recessive disease, the energy-depleted erythrocytes are prone to hemolysis. Additionally, the ATP-deficiency in the erythroblasts leads to ineffective erythropoiesis. Patients with PK deficiency have symptoms and signs due to ineffective erythropoiesis and chronic hemolysis [29] and the clinical picture resembles NTDT.
The results showed a significant decrease of reactive oxygen species levels and an improvement of ineffective erythropoiesis. Moreover, a reduction of liver iron deposition and an increase of hepcidin levels were observed [30]. The interim results from a phase 2 trial (NCT03692052) of mitapivat in 13 adults with NTDT (α- or β-thalassemia) were presented at the 2020 ASH annual meeting. During the period of 4–12 weeks, the mean Hb increase from baseline was 1.34 g/dL with concomitant amelioration of hemolysis indices.
The alteration of intracellular cyclic guanosine monophosphate (cGMP) is a novel therapeutic objective for sickle cell disease and thalassemia. The cGMP-dependent pathway is significant for the production of HbF and has multiple roles in vascular biology. As phosphodiesterase (PDE) 9 selectively degrades cGMP in erythropoietic cells, the use of inhibitors of PDE9 can result in increased cGMP levels and the reactivation of HbF [31][32].
IMR-687 is a novel agent that has been developed for the inhibition of PDE 9. The oral use of IMR-687 in sickle cell disease patients has been recently completed (NCT03401112) and has shown to stimulate HbF production and to improve Hb levels and hemolysis indices [33]. A similar phase 2 study has been launched in order to evaluate the safety and tolerability of IMR-687 given once daily for 36 weeks in TDT and NTDT adult thalassemic patients (NCT04411082).
Hepcidin, a protein produced from the hepatocytes, is the key controller of iron metabolism. Hepcidin degrades ferroportin, the main iron exporter in intestinal cells; macrophages of the reticuloendothelial system; and the hepatocytes. In β-thalassemia, hepcidin production is inhibited through the action of ERFE, contributing to increased intestinal iron absorption and tissue deposition [6]. Restoration of hepcidin levels could improve iron overload and ineffective erythropoiesis [34].
Hepcidin production is mainly regulated by matriptase-2 (MT-2), a transmembrane serine protease, which is encoded by theTMPRSS6gene. MT-2 inhibits hepcidin activation by cleaving membrane hemojuvelin [35]. In mouse models with NTDT, the inhibition ofTMPRSS6with silencing RNAs or antisense oligonucleotides caused a rise of hepcidin and an amelioration of anemia and iron deposition [36][37][38][39]. Phase 2 clinical trials are already recruiting NTDT patients in order to assess the efficacy, safety, and pharmacokinetics of the short interfering RNAs, SLN124 (NCT04718844), and IONIS
The use of minihepcidins, which act as hepcidin agonists, has been proven to improve ineffective erythropoiesis and splenomegaly in a TDT mouse model [40]. Recent initial results of a phase 2 trial (NCT04054921) on the subcutaneous use of PTG-300, a hepcidin mimetic analog, have shown a reduction in serum iron parameters and a 20% or greater reduction in transfusions, with mild adverse effects [41]. Another approach is to restrict the availability of iron, targeting both the iron overload and the ineffective erythropoiesis, with the use of ferroportin inhibitors (VIT-2763). A phase 1 study in healthy volunteers has shown that the drug was well accepted with no significant safety concerns along with a transient decrease in mean serum iron levels and transferrin saturation [42].
4. Conclusions—Key Points
In β-thalassemia, the globin chain inequality, ineffective erythropoiesis, and iron tissue deposition are the main underlying pathophysiological mechanisms that lead to the complications of the disease. Recent research on these mechanisms has revealed novel curative and “disease-modifying” therapeutic approaches.
Clinically meaningful progress in the techniques of allo-HSCT has been accomplished mainly in the area of the conditioning regimen and donor selection. Gene manipulation studies have exhibited remarkable advances. The need for a specialized center for the procedure as well as the high financial cost are barriers to the widespread application of gene therapy. However, gene therapy has more a measurable and objective effect than luspatercept and may lead to transfusion independence.
A very recent and highly sophisticated approach is the application of epigenetic and genomic editing methods that target the silencing of γ-globin repressors, such as BCL11A, or the correction of β-globin gene. Remarkable results have been reported in preclinical studies, while the preliminary results from clinical trials are more than promising.
It is a pharmacological therapy that can be an additive to standard care with manageable side effects that allows the patient to discontinue the drug when needed or in cases of poor response, whereas this in not realistic in gene therapy. It can also be an alternative option in cases of young individuals above 18 years of age who do not have the criteria for transplantation or the availability of a matching related donor. The use of luspatercept is also being studied in NTDT patients, and clinical trials have been designed for children. Furthermore, the fact that luspatercept reduces the need for transfusions will be beneficial in health care systems that have a shortage of blood products, especially during this period of the SARS-CoV-2 pandemic.
The oral administration of mitapivat in a small number of patients has shown promising results in ongoing clinical trials. An alternative way to improve anemia in thalassemic patients is to stimulate HbF production with the use of phosphodiesterase 9 inhibitors, such as IMR-687. This agent has shown encouraging results in sickle cell disease patients and is currently being tested in aduts with TDT and NTDT. A different therapeutic approach is to target the dysregulation of iron homeostasis and indirectly improve erythropoiesis, by using hepcidin agonists (inhibitors of TMPRSS6 and minihepcidins) or ferroportin inhibitors (VIT-2763).
For individuals with NTDT, transfusions are given as needed in certain clinical settings. In TDT, the mainstream treatment is chronic transfusions to alleviate anemia and suppress extramedullary hematopoiesis, with parallel management of the excess free iron and the complications of the disease. Matched related transplantation is the preferable approach, but other alternatives can be considered in terms of donor selection and conditioning regimens. In the case of a non-suitable donor, gene therapy with β-gene addition is an option in certified transplant centers.
References
- Rund, D.; Rachmilewitz, E. Beta-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146.
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844.
- Blank, U.; Karlsson, S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia 2011, 25, 1379–1388.
- Gardenghi, S.; Marongiu, M.F.; Ramos, P.; Guy, E.; Breda, L.; Chadburn, A.; Liu, Y.; Amariglio, N.; Rechavi, G.; Rachmilewitz, E.A.; et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007, 109, 5027–5035.
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684.
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037.
- Origa, R.; Cazzola, M.; Mereu, E.; Danjou, F.; Barella, S.; Giagu, N.; Galanello, R.; Swinkels, D.W. Differences in the erythropoiesis-hepcidin-iron store axis between hemoglobin H disease and beta-thalassemia intermedia. Haematologica 2015, 100, e169–e171.
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101.
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186.
- Lucarelli, G.; Isgro, A.; Sodani, P.; Gaziev, J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb. Perspect. Med. 2012, 2, a011825.
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493.
- Lal, A.; Locatelli, F.; Kwiatkowski, J.L.; Kulozik, A.E.; Yannaki, E.; Porter, J.B.; Thuret, I.; Sauer, M.G.; Elliot, H.; Chen, Y.; et al. Northstar-3: Interim Results from a Phase 3 Study Evaluating Lentiglobin Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia and Either a β0 or IVS-I-110 Mutation at Both Alleles of the HBB Gene. Blood 2019, 134, 815.
- Kwiatkowski, J.L.; Walters, M.C.; Hongeng, S.; Locatelli, F.; Rasko, J.E.L.; Cavazzana, M.; Chen, Y.; Colvin, R.A.; Thompson, A.A. Long-Term Efficacy and Safety of Betibeglogene Autotemcel Gene Therapy for the Treatment of Transfusion-Dependent β-Thalassemia: Results in Patients with up to 6 Years of Follow-up. In Proceedings of the 62nd ASH Annual Meeting, San Diego, CA, USA, 5–8 December 2020.
- Thompson, A.A.; Kwiatkowski, J.L.; Porter, J.B.; Hongeng, S.; Yannaki, E.; Kulozik, A.E.; Sauer, M.G.; Thrasher, A.J.; Thuret, I.; Lal, A.; et al. Favorable Outcomes in Pediatric Patients in the Phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) Studies of Betibeglogene Autotemcel Gene Therapy for the Treatment of Transfusion-Dependent β-Thalassemia. In Proceedings of the 62nd ASH Annual Meeting, San Diego, CA, USA, 5–8 December 2020.
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ss-thalassemia. Nat. Med. 2019, 25, 234–241.
- Guda, S.; Brendel, C.; Renella, R.; Du, P.; Bauer, D.E.; Canver, M.C.; Grenier, J.K.; Grimson, A.W.; Kamran, S.C.; Thornton, J.; et al. miRNA-embedded shRNAs for Lineage-specific BCL11A Knockdown and Hemoglobin F Induction. Mol. Ther. 2015, 23, 1465–1474.
- Smith, A.R.; Schiller, G.J.; Vercellotti, G.M.; Kwiatkowski, J.L.; Krishnamurti, L.; Esrick, E.B.; Williams, D.A.; Miller, W.P.; Woolfson, A.; Walters, M.C. Preliminary Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-Dependent Beta Thalassemia. Blood 2019, 134, 3544.
- Hinck, A.P. Structural studies of the TGF-betas and their receptors—Insights into evolution of the TGF-beta superfamily. FEBS Lett. 2012, 586, 1860–1870.
- Yuki, R.; Tatewaki, T.; Yamaguchi, N.; Aoyama, K.; Honda, T.; Kubota, S.; Morii, M.; Manabe, I.; Kuga, T.; Tomonaga, T.; et al. Desuppression of TGF-beta signaling via nuclear c-Abl-mediated phosphorylation of TIF1gamma/TRIM33 at Tyr-524, -610, and -1048. Oncogene 2019, 38, 637–655.
- Sako, D.; Grinberg, A.V.; Liu, J.; Davies, M.V.; Castonguay, R.; Maniatis, S.; Andreucci, A.J.; Pobre, E.G.; Tomkinson, K.N.; Monnell, T.E.; et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 2010, 285, 21037–21048.
- Suragani, R.N.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg, A.V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood 2014, 123, 3864–3872.
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat. Med. 2014, 20, 398–407.
- Martinez, P.; Bhasin, M.; Li, R.; Pearsall, S.; Kumar, R.; Suragani, R. RAP-536 (murine analog of ACE-536/Luspatercept) inhibits SMAD2/3 signaling and promotes erythroid diffentiation by restoring GATA-1 function in a murine model of b-thalassemia. In Proceedings of the 21st Congress of the European Haematology Association, Copenhagen, Denmark, 9–12 June 2016.
- Martinez, P.A.; Li, R.; Ramanathan, H.N.; Bhasin, M.; Pearsall, R.S.; Kumar, R.; Suragani, R. Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine beta-thalassaemia by increasing GATA-1 availability. J. Cell. Mol. Med. 2020, 24, 6162–6177.
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with beta-thalassemia. Blood 2019, 133, 1279–1289.
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galacteros, F.; Taher, A.T.; Arlet, J.B.; Ribeil, J.A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor beta ligand trap, improves anemia in beta-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484.
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent beta-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231.
- Piga, A. Transfusion Dependent Beta (β)-Thalassemia (BEYOND). Available online: (accessed on 8 May 2021).
- Aizawa, S.; Harada, T.; Kanbe, E.; Tsuboi, I.; Aisaki, K.; Fujii, H.; Kanno, H. Ineffective erythropoiesis in mutant mice with deficient pyruvate kinase activity. Exp. Hematol. 2005, 33, 1292–1298.
- Matte, A.; Beneduce, E.; Siciliano, A.; Kosinski, P.; Janin, A.; Lebouef, C.; Iolacson, A.; De Falco, L.; Dang, L.; Kung, C.; et al. The pyruvate kinase activator AG-348 improves murine b-thalassemic anemia and corrects ineffective erythropoiesis. In Proceedings of the 21st Congress of the European Haematology Association, Copenhagen, Denmark, 9–12 June 2016.
- Almeida, C.B.; Traina, F.; Lanaro, C.; Canalli, A.A.; Saad, S.T.; Costa, F.F.; Conran, N. High expression of the cGMP-specific phosphodiesterase, PDE9A, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br. J. Haematol. 2008, 142, 836–844.
- McArthur, J.G.; Svenstrup, N.; Chen, C.; Fricot, A.; Carvalho, C.; Nguyen, J.; Nguyen, P.; Parachikova, A.; Abdulla, F.; Vercellotti, G.M.; et al. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica 2020, 105, 623–631.
- Bronte-Hall, L.; Andemariam, B.; Gershwin, B.; Lugthart, S.; Mant, T.; Howard, J.; Fok, H.; Eleftheriou, P.; Hagar, R.W.; Mason, J.; et al. Benefits and Safety of Long-Term Use of IMR-687 As Monotherapy or in Combination with a Stable Dose of Hydroxyurea (HU) in 2 Adult Sickle Cell Patients. Blood 2020, 136, 29–30.
- Schmidt, P.J.; Fleming, M.D. Modulation of hepcidin as therapy for primary and secondary iron overload disorders: Preclinical models and approaches. Hematol./Oncol. Clin. N. Am. 2014, 28, 387–401.
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511.
- Casu, C.; Aghajan, M.; Oikonomidou, P.R.; Guo, S.; Monia, B.P.; Rivella, S. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica 2016, 101, e8–e11.
- Schmidt, P.J.; Racie, T.; Westerman, M.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am. J. Hematol. 2015, 90, 310–313.
- Stagg, D.B.; Whittlesey, R.L.; Li, X.; Lozovatsky, L.; Gardenghi, S.; Rivella, S.; Finberg, K.E. Genetic loss of Tmprss6 alters terminal erythroid differentiation in a mouse model of beta-thalassemia intermedia. Haematologica 2019, 104, e442–e446.
- Schmidt, P.J.; Liu, K.; Visner, G.; Fitzgerald, K.; Fishman, S.; Racie, T.; Hettinger, J.L.; Butler, J.S.; Fleming, M.D. RNAi-mediated reduction of hepatic Tmprss6 diminishes anemia and secondary iron overload in a splenectomized mouse model of beta-thalassemia intermedia. Am. J. Hematol. 2018, 93, 745–750.
- Casu, C.; Chessa, R.; Liu, A.; Gupta, R.; Drakesmith, H.; Fleming, R.; Ginzburg, Y.Z.; MacDonald, B.; Rivella, S. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult beta-thalassemia major. Haematologica 2020, 105, 1835–1844.
- Lai, A.; Voskaridou, E.; Flevari, P.; Taher, A.; Chew, L.; Valone, F.; Gupta, S.; Viprakasit, V. A hepcidin mimetic, PTG-300, demonstrates, pharmacodynamic effects indicating reduced iron availability in transfusion-dependent beta-thalassemia subjects. In Proceedings of the 25th EHA Congress, virtual, 11–21 June 2020.
- Richard, F.; van Lier, J.J.; Roubert, B.; Haboubi, T.; Gohring, U.M.; Durrenberger, F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am. J. Hematol. 2020, 95, 68–77.


