 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiaoqiang Huang | + 2096 word(s) | 2096 | 2021-07-08 12:29:28 | | | |
| 2 | Amina Yu | Meta information modification | 2096 | 2021-07-12 06:24:31 | | |
Video Upload Options
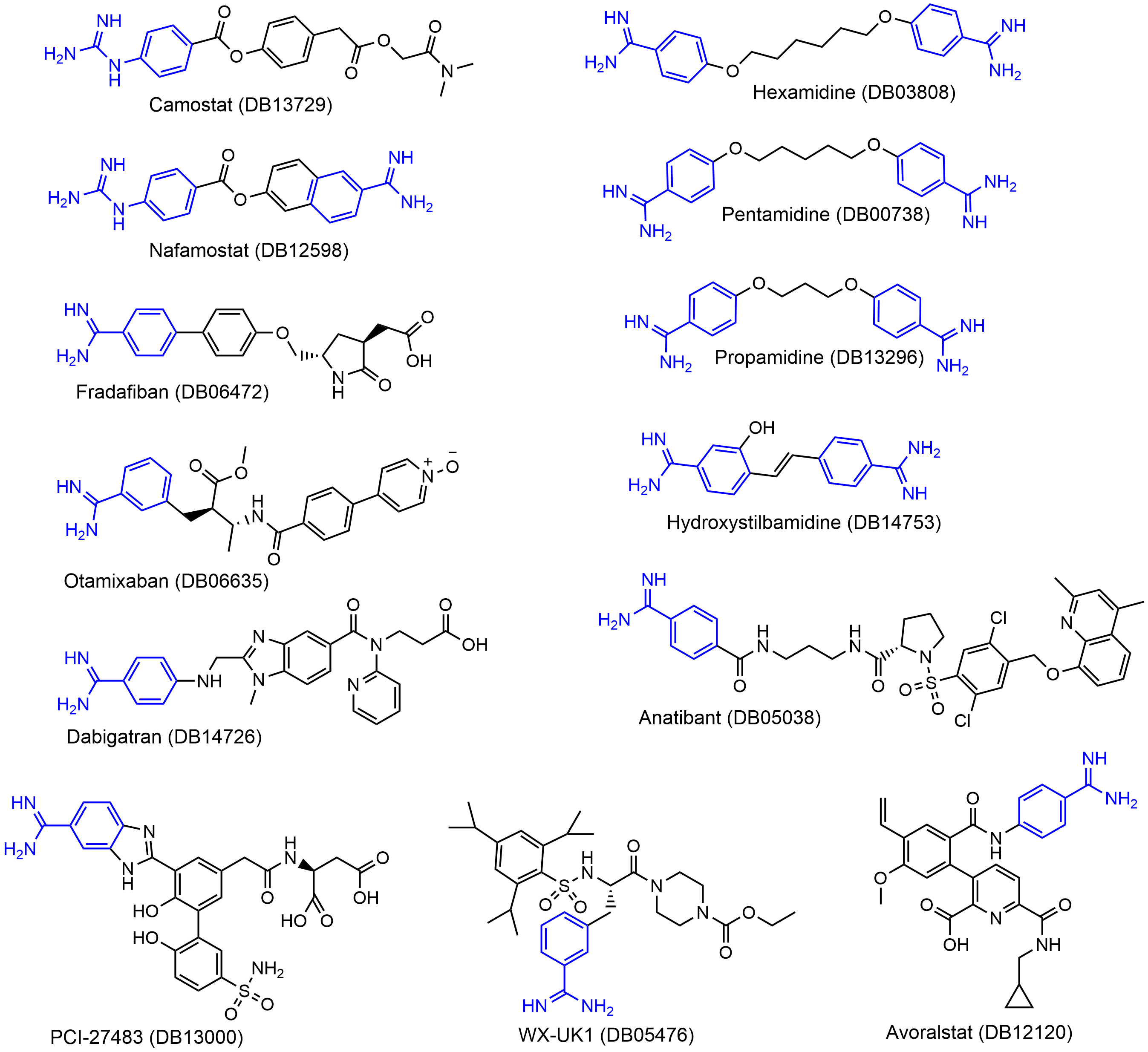
We identified a set of 13 approved or clinically investigational drugs with positively charged guanidinobenzoyl and/or aminidinobenzoyl groups, including the experimentally verified TMPRSS2 inhibitors Camostat and Nafamostat. Molecular docking suggested that the guanidinobenzoyl or aminidinobenzoyl group in all the drugs could form putative salt bridge interactions with the side-chain carboxyl group of Asp435 located in the S1 pocket of TMPRSS2. Molecular dynamics simulations further revealed the high stability of the putative salt bridge interactions over long-time simulations. These results suggest that the proposed compounds, in addition to Camostat and Nafamostat, could be effective TMPRSS2 inhibitors for COVID-19 treatment by occupying the S1 pocket with the hallmark positively charged groups.
1. Introduction
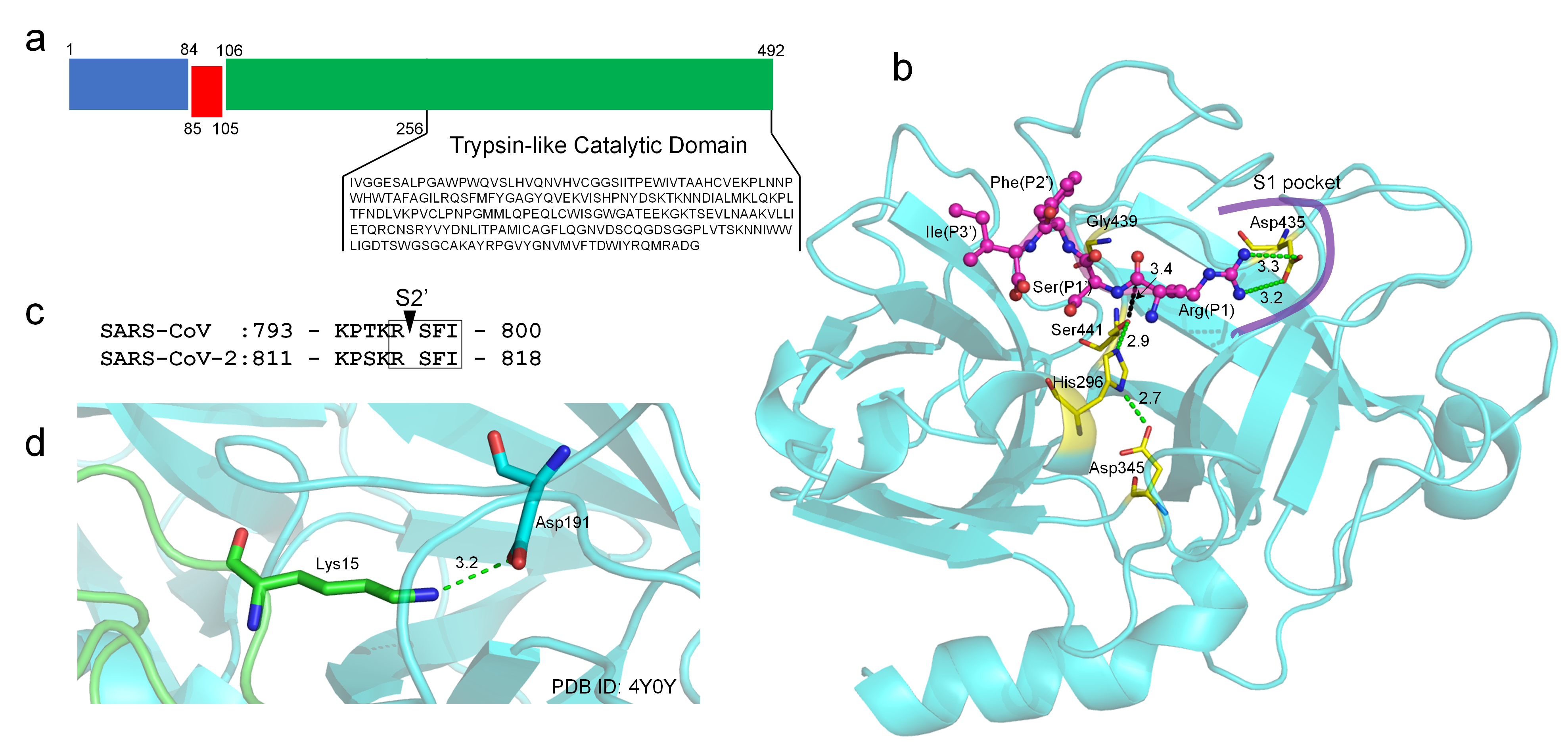
2. TMPRSS2 Sequence and Structural Model
3. Guanidinobenzoyl- or Aminidinobenzoyl-Containing Drugs
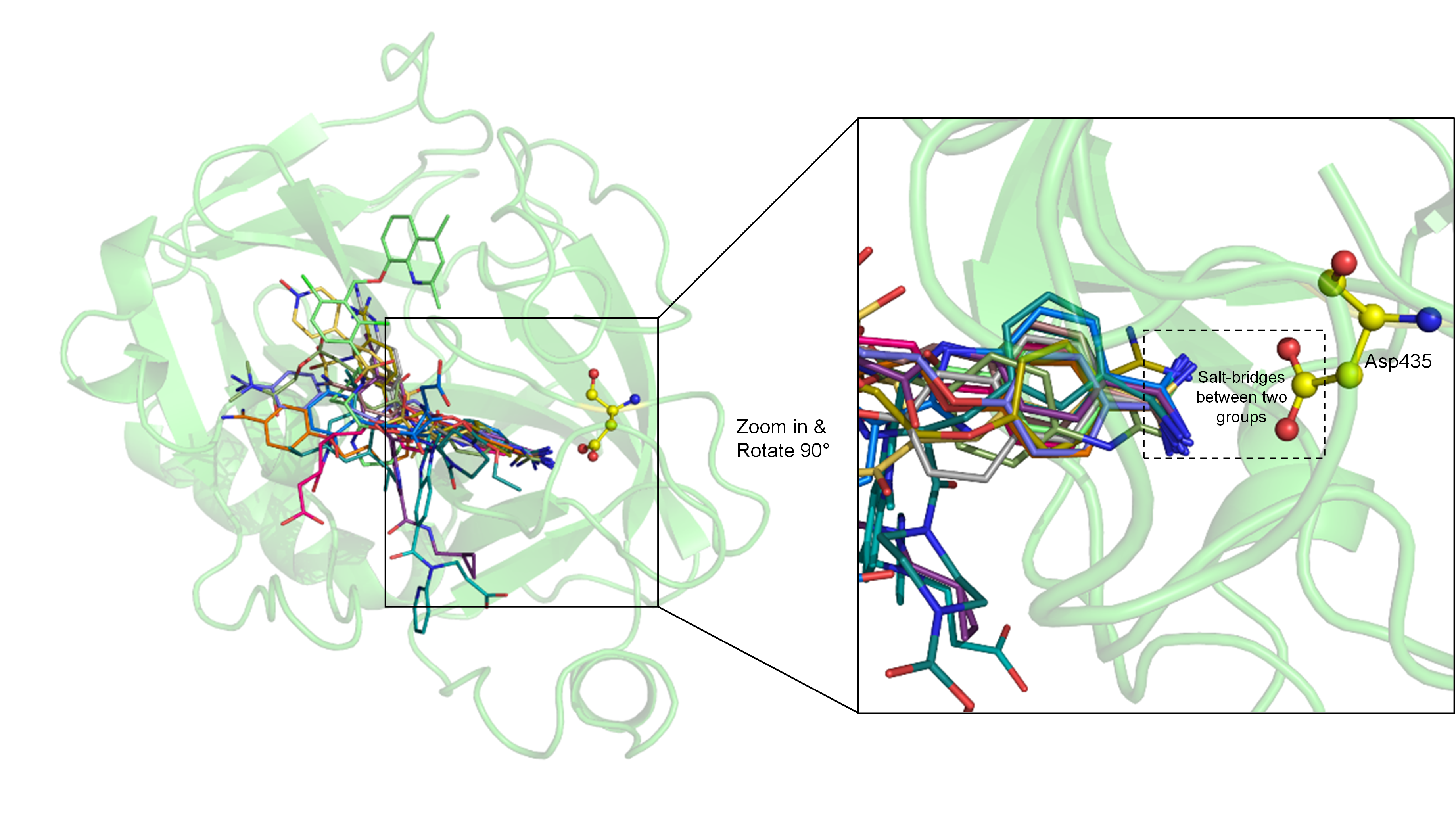
4. Molecular Docking Suggests Salt Bridge Interactions between Guanidinobenzoyl or Aminidinobenzoyl and Asp435
5. MD Simulations Reveal High Stability of the Putative Salt Bridge Interactions
We carried out MD simulations to examine the binding stability between TMPRSS2 and the drugs. Before MD, the top ten poses (if they existed) ranked by EvoEF2 for each drug were parameterized using the ACPYPE[20] program with the AM1-BCC[21] charge model. The number of poses that can be successfully applied to MD for the 13 drugs were 10 (DB00738), 10 (DB03808), 10 (DB05038), 9 (DB05476), 8 (DB06472), 10 (DB06635), 7 (DB12120), 6 (DB12598), 5 (DB13000), 10 (DB13296), 10 (DB13729), 10 (DB14726), and 4 (DB14753), respectively. TMPRSS2 in complex with each suitable drug pose was subjected to a long-time (100 ns) MD simulation using GROMACS v2020.4[22].
According to docking models, the guanidinobenzoyl or aminidinobenzoyl groups were docked into the deep S1 pocket and formed salt bridge interactions with Asp435. We examined the stability of the putative salt bridge interactions by measuring the minimum distance between the positively charged guanidinobenzoyl or aminidinobenzoyl group and the negatively charged carboxyl group of Asp435 (denoted as \( d_{ON}^{min} \)); only the distances between the nitrogen and oxygen atoms were calculated. All the drugs had at least one pose with a mean and median \( d_{ON}^{min} \) fluctuating around 2.8 Å, an ideal salt bridge distance, with small deviations (Figure 4). Therefore, a long-time MD simulation indicated the high stability of the putative salt bridge interactions between the guanidinobenzoyl or aminidinobenzoyl group and Asp435, which should be important for TMPRSS2 inhibition.
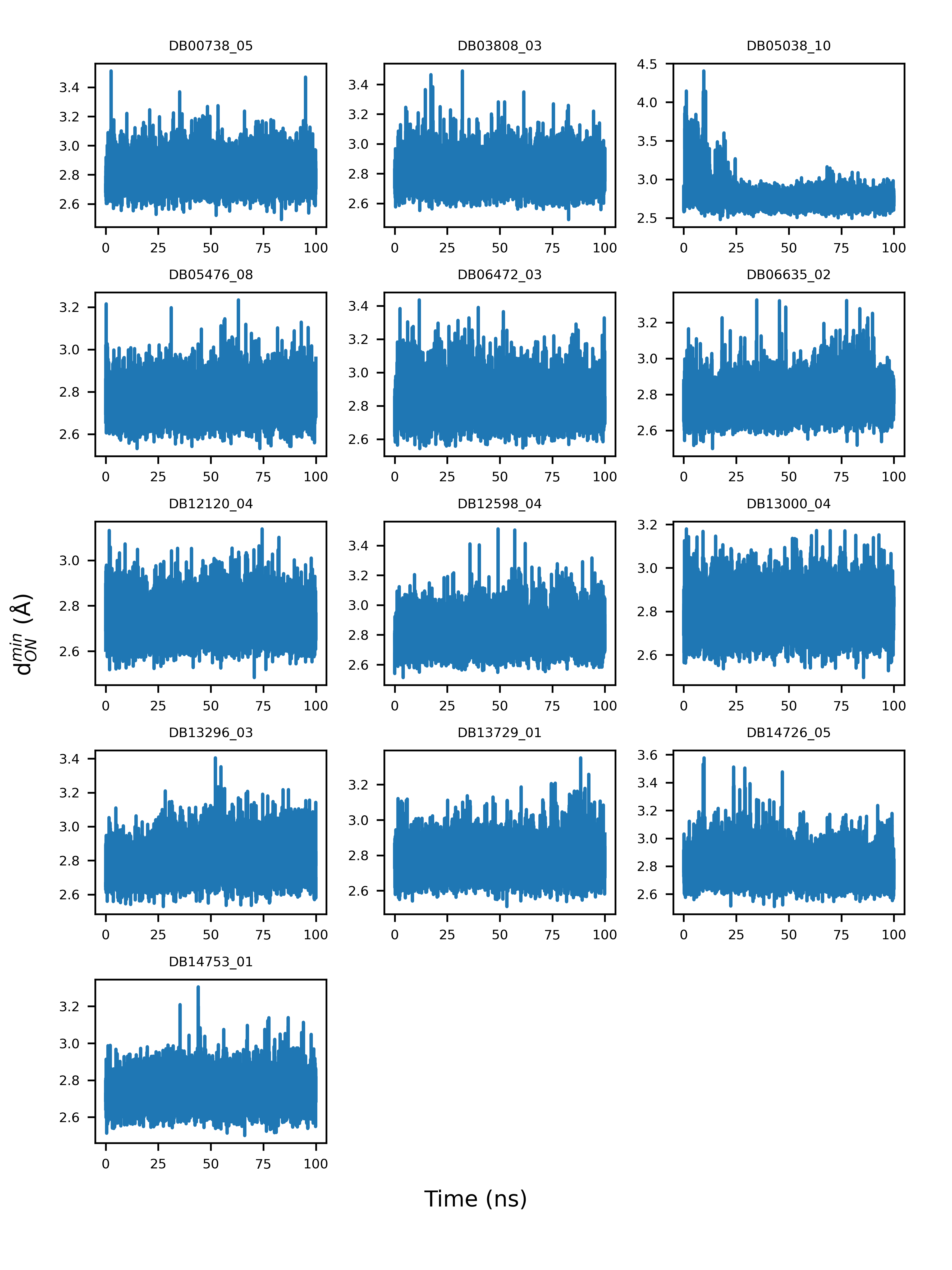 Figure 4. Example illustration of the variations of the minimum distance between guanidinobenzoyl or aminidinobenzoyl and the Asp435 carboxyl (\( d_{ON}^{min} \)) over a 100-ns simulation time.
Figure 4. Example illustration of the variations of the minimum distance between guanidinobenzoyl or aminidinobenzoyl and the Asp435 carboxyl (\( d_{ON}^{min} \)) over a 100-ns simulation time.
6. Conclusions
Building on the recent finding that the positively charged groups in Camostat and Nafamostat play a critical role in inhibiting TMPRSS2 by stable binding with the conserved aspartic acid Asp435 in the S1 pocket of TMPRSS2, we identified a narrowed set of 13 compounds (three FDA-approved and 10 investigational drugs) with positively charged guanidinobenzoyl or aminidinobenzoyl groups and computationally assessed their potency for inhibiting TMPRSS2. This work differed from virtual screening studies that focus on identifying TMPRSS2 inhibitors from huge drug databases. Usually, a virtual screening study suggests a long list of candidates for experimental tests but, finally, comes up with few positive hits; instead, here, we tried to evaluate and repurpose only a few very promising candidates. The molecular docking studies showed that all the 13 drugs indeed utilized guanidinobenzoyl or aminidinobenzoyl to form favorable salt bridge interactions with the Asp435 carboxyl, and a series of long-time (100 ns) MD simulations revealed the high stability of the salt bridge interactions between each drug and TMPRSS2, although each whole ligand may undergo large conformational changes. Collectively, the computational data supported these drugs as potential TMPRSS2 inhibitors for treating COVID-19.
References
- Rolando Cannalire; Irina Stefanelli; Carmen Cerchia; Andrea R. Beccari; Sveva Pelliccia; Vincenzo Summa; SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. International Journal of Molecular Sciences 2020, 21, 5707, 10.3390/ijms21165707.
- Konrad H. Stopsack; Lorelei A. Mucci; Emmanuel S. Antonarakis; Peter S. Nelson; Philip W. Kantoff; TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention?. Cancer Discovery 2020, 10, 779-782, 10.1158/2159-8290.cd-20-0451.
- Markus Hoffmann; Hannah Kleine-Weber; Simon Schroeder; Nadine Krüger; Tanja Herrler; Sandra Erichsen; Tobias S. Schiergens; Georg Herrler; Nai-Huei Wu; Andreas Nitsche; et al.Marcel A. MüllerChristian DrostenStefan Pöhlmann SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271-280.e8, 10.1016/j.cell.2020.02.052.
- Markus Hoffmann; Heike Hofmann-Winkler; Joan C. Smith; Nadine Krüger; Prerna Arora; Lambert K. Sørensen; Ole S. Søgaard; Jørgen Bo Hasselstrøm; Michael Winkler; Tim Hempel; et al.Lluís RaichSimon OlssonOlga DanovDanny JonigkTakashi YamazoeKatsura YamatsutaHirotaka MizunoStephan LudwigFrank NoéMads KjolbyArmin BraunJason M. SheltzerStefan Pöhlmann Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255, 10.1016/j.ebiom.2021.103255.
- Markus Hoffmann; Simon Schroeder; Hannah Kleine-Weber; Marcel A. Müller; Christian Drosten; Stefan Pöhlmann; Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrobial Agents and Chemotherapy 2020, 64, e00754-20, 10.1128/aac.00754-20.
- Jonathan H. Shrimp; Stephen Kales; Philip E. Sanderson; Anton Simeonov; Min Shen; Matthew D. Hall; An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacology & Translational Science 2020, 3, 997-1007, 10.1021/acsptsci.0c00106.
- Mizuki Yamamoto; Maki Kiso; Yuko Sakai-Tagawa; Kiyoko Iwatsuki-Horimoto; Masaki Imai; Makoto Takeda; Noriko Kinoshita; Norio Ohmagari; Jin Gohda; Kentaro Semba; et al.Zene MatsudaYasushi KawaguchiYoshihiro KawaokaJun-Ichiro Inoue The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629, 10.3390/v12060629.
- Tim Hempel; Lluís Raich; Simon Olsson; Nurit P. Azouz; Andrea M. Klingler; Markus Hoffmann; Stefan Pöhlmann; Marc E. Rothenberg; Frank Noé; Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chemical Science 2020, 12, 983-992, 10.1039/d0sc05064d.
- Wei Zheng; Yang Li; Chengxin Zhang; Robin Pearce; S. M. Mortuza; Yang Zhang; Deep‐learning contact‐map guided protein structure prediction in CASP13. Proteins: Structure, Function, and Bioinformatics 2019, 87, 1149-1164, 10.1002/prot.25792.
- Yang Zhang; I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 1-8, 10.1186/1471-2105-9-40.
- Yang Zhang; Jeffrey Skolnick; Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics 2004, 57, 702-710, 10.1002/prot.20264.
- Vincent Chen; W. Bryan Arendall; Jeffrey J. Headd; Daniel Keedy; Robert M. Immormino; Gary J. Kapral; Laura W. Murray; Jane Richardson; David C. Richardson; MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography 2009, 66, 12-21, 10.1107/s0907444909042073.
- Pei Zhou; Bowen Jin; Hao Li; Sheng-You Huang; HPEPDOCK: a web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic Acids Research 2018, 46, W443-W450, 10.1093/nar/gky357.
- David J. Huggins; Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. Journal of Molecular Graphics and Modelling 2020, 100, 107710-107710, 10.1016/j.jmgm.2020.107710.
- Diego E. Escalante; David M. Ferguson; Structural modeling and analysis of the SARS-CoV-2 cell entry inhibitor camostat bound to the trypsin-like protease TMPRSS2. Medicinal Chemistry Research 2021, 30, 399-409, 10.1007/s00044-021-02708-7.
- Alastair D. G. Lawson; Malcolm MacCoss; Jag P. Heer; Importance of Rigidity in Designing Small Molecule Drugs To Tackle Protein–Protein Interactions (PPIs) through Stabilization of Desired Conformers. Journal of Medicinal Chemistry 2017, 61, 4283-4289, 10.1021/acs.jmedchem.7b01120.
- Christophe Verlinde; Wim Gj Hol; Structure-based drug design: progress, results and challenges. Structure 1994, 2, 577-587, 10.1016/s0969-2126(00)00060-5.
- Na Zhang; Hongtao Zhao; Enriching screening libraries with bioactive fragment space. Bioorganic & Medicinal Chemistry Letters 2016, 26, 3594-3597, 10.1016/j.bmcl.2016.06.013.
- Xiaoqiang Huang; Robin Pearce; Yang Zhang; EvoEF2: accurate and fast energy function for computational protein design. Bioinformatics 2019, 36, 1135-1142, 10.1093/bioinformatics/btz740.
- Alan W Sousa Da Silva; Wim F Vranken; ACPYPE - AnteChamber PYthon Parser interfacE. BMC Research Notes 2011, 5, 367-367, 10.1186/1756-0500-5-367.
- Araz Jakalian; Bruce L. Bush; David B. Jack; Christopher I. Bayly; Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. Journal of Computational Chemistry 2000, 21, 132-146, 10.1002/(sici)1096-987x(20000130)21:2<132::aid-jcc5>3.0.co;2-p.
- Mark James Abraham; Teemu Murtola; Roland Schulz; Szilárd Páll; Jeremy Smith; Berk Hess; Erik Lindahl; GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19-25, 10.1016/j.softx.2015.06.001.


