 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristiano Capurso | + 4687 word(s) | 4687 | 2021-07-07 05:47:35 | | | |
| 2 | Peter Tang | Meta information modification | 4687 | 2021-07-12 04:04:02 | | |
Video Upload Options
Aging is physiologically characterized by a decrease in lean mass, bone mineral density and, to a lesser extent, fat mass. The onset of sarcopenia leads to weakness and a further decrease in physical activity. An insufficient protein intake, which we often observe in patients of advanced age, certainly accelerates the progression of sarcopenia. In addition, many other factors (e.g., insulin resistance, impaired protein digestion and absorption of amino acids) reduce the stimulation of muscle protein synthesis in the elderly, even if the protein intake is adequate. Inadequate intake of foods can also cause micronutrient deficiencies that contribute to the development of frailty. The Mediterranean diet is recognized to be a “healthy food” dietary pattern; high adherence to this dietary pattern is associated with a lower incidence of chronic diseases and lower physical impairment in old age.
1. Introduction
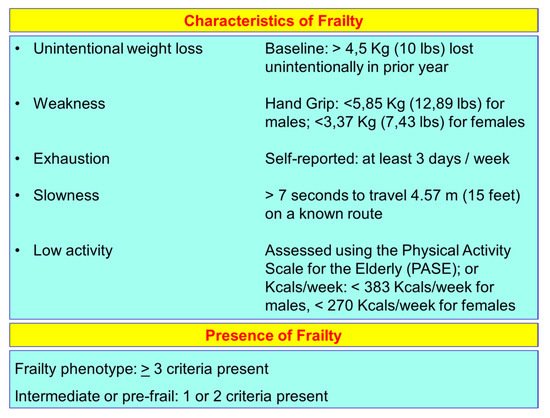
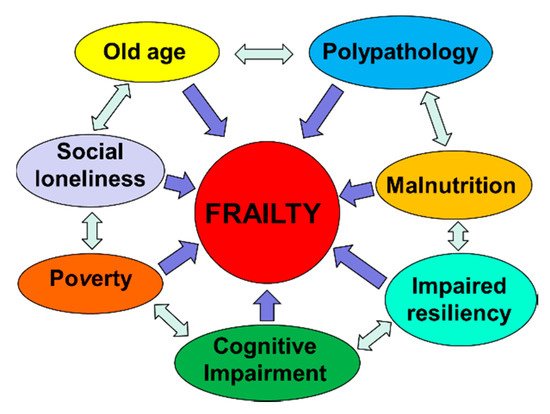
2. Aging and Frailty

3. Aging, or Cellular Senescence, and Health
4. The Role of Senescence in the Progression of Diabetes Mellitus and Atherosclerosis
5. Caloric Restriction, Effects on Metabolism of Adipose Tissue and Increase of Longevity
6. Caloric Restriction and Inflammatory State
7. Caloric Restriction, Mitochondria Activity and Reactive Oxygen Species Production
8. Caloric Restriction, Hormesis and Mitochondria Activity during Aging
9. Caloric Restriction and DNA Methylation
10. Caloric Restriction, Metabolic Adaptation and Oxidative Damage
11. Mediterranean Diet, Cardiovascular Disease and Mortality
|
Author and Year of Publication |
Study Design |
Sample Size |
Risk of Mortality |
|---|---|---|---|
|
Trichopoulou, 2003, [84] |
Population-based, prospective study |
8895 men and 13,148 women |
Death from any cause: HR = 0.75 (95% CI: 0.64–0.87) for a Two-Point Increase in the Mediterranean-Diet Score Death from coronary heart disease: HR = 0.67 (95% CI: 0.47–0.94) for a Two-Point Increase in the Mediterranean-Diet Score Death from cancer: HR = 0.76 (95% CI: 0.59–0.98) for a Two-Point Increase in the Mediterranean-Diet Score |
|
Estruch, 2013, [86] |
Parallel-group, multicentre, randomized trial |
1050 men and 1493 women with MD with EVOO 1128 men and 1326 women with MD with nuts 987 men and 1463 women with Control Diet |
Myocardial infarction, stroke, and death from cardiovascular causes: HR = 0.70 (95% CI: 0.54–0.92, p = 0.01) for MD with EVOO vs. Control Diet HR = 0.72 (95% CI: 0.54–0.96, p = 0.03) for MD with Nuts vs. Control Diet Death from any cause: HR = 0.82 (95% CI: 0.64–1.07, p = 0.15) for MD with EVOO vs. Control Diet HR = 0.97 (95% CI: 0.74–1.26, p = 0.82) for MD with Nuts vs. Control Diet |
|
Estruch, 2018, [87] |
Parallel-group, multicentre, randomized trial |
1050 men and 1493 women with MD with EVOO 1128 men and 1326 women with MD with nuts 987 men and 1463 women with Control Diet |
Myocardial infarction: HR = 0.82 (95% CI: 0.52–1.30) for MD with EVOO vs. Control Diet HR = 0.76 (95% CI: 0.47–1.25) for MD with Nuts vs. Control Diet Stroke: HR = 0.65 (95% CI: 0.44–0.95) for MD with EVOO vs. Control Diet HR = 0.54 (95% CI: 0.35–0.82) for MD with Nuts vs. Control Diet Death from cardiovascular causes: HR = 0.62 (95% CI: 0.36–1.06) for MD with EVOO vs. Control Diet HR = 1.02 (95% CI: 0.63–1.67) for MD with Nuts vs. Control Diet Death from any cause: HR = 0.90 (95% CI: 0.69–1.18) for MD with EVOO vs. Control Diet HR = 1.12 (95% CI: 0.86–1.47) for MD with Nuts vs. Control Diet |
|
Sofi, 2008, [88] |
Meta-analysis of prospective cohort studies |
1,574,299 subjects from 12 studies |
Mortality from cardiovascular diseases: RR = 0.91 (95% CI: 0.87–0.95) Mortality from any cause: RR = 0.91 (95% CI: 0.89–0.94 Mortality from cancer: RR = 0.94 (95% CI: 0.92–0.96) Incidence of Parkinson’s disease and Alzheimer’s disease: RR = 0.87 (95% CI: 0.80–0.96) |
|
Sofi, 2010, [89] |
Meta-analysis of prospective cohort studies |
508,393 subjects from 7 studies |
Mortality from cardiovascular diseases: RR = 0.90 (95% CI: 0.87–0.93) Mortality from any cause: RR = 0.92 (95% CI: 0.90–0.94) Mortality from cancer: RR = 0.94 (95% CI: 0.92–0.96) Incidence of neurodegenerative disease: RR = 0.87 (95% CI: 0.81–0.94) |
|
Kromhout, 2018, [92] |
Prospective Cohort Study |
12,763 subjects from 16 cohorts of the Seven Countries Study |
Mortality from cardiovascular diseases: Inverse association between consumption of cereals, vegetables, legumes, and alcohol and long-term CHD mortality rates (r = −0.52 to −0.62) Positive association between consumption of hard fat plus sweet products, animal foods except fish, and long-term CHD mortality rates (r = 0.68 to 0.84) |
References
- Population Structure and Ageing. Available online: (accessed on 1 June 2019).
- Kinsella, K.; Phillips, D.R. Global Aging: The Challenge of Success; Population Bulletin; Population Reference Bureau: Washington, DC, USA, 2005; Volume 60.
- United Nations. The World at Six Billion. Available online: (accessed on 1 June 2019).
- Beard, J.R. The World report on ageing and health: A policy framework for healthy ageing. Lancet 2016, 387, 2145–2154.
- Beard, J.R.; Bloom, D.E. Towards a comprehensive public health response to population ageing. Lancet 2015, 385, 658–661.
- Age Wave, Sun America. Age Wave/Sun America Retirement Reset Study; Age Wave, Sun America: Los Angeles, CA, USA, 2011.
- Britton, A.; Shipley, M.; Singh-Manoux, A.; Marmot, M.G. Successful aging: The contribution of early life and midlife risk factors. J. Am. Geriatr. Soc. 2008, 56, 1098–1105.
- Akbaraly, T.; Sabia, S.; Hagger-Johnson, G.; Tabak, A.G.; Shipley, M.J.; Jokela, M.; Brunner, E.J.; Hamer, M.; Batty, G.D.; Singh-Manoux, A.; et al. Does overall diet in midlife predict future aging phenotypes? A cohort study. Am. J. Med. 2013, 126, 411–419.
- Samieri, C.; Sun, Q.; Townsend, M.K.; Chiuve, S.E.; Okereke, O.I.; Willett, W.C.; Stampfer, M.; Grodstein, F. The association between dietary patterns at midlife and health in aging an observational study. Ann. Intern. Med. 2013, 159, 584–591.
- Walston, J.; Hadley, E.C.; Ferrucci, L.; Guralnik, J.M.; Newman, A.B.; Studenski, S.A.; Ershler, W.B.; Harris, T.; Fried, L.P. Research agenda for frailty in older adults: Toward a better understanding of physiology and aetiology: Summary from the American Geriatrics Society/National Institute on Aging Research Conference on Frailty in Older Adults. J. Am. Geriatr. Soc. 2006, 54, 991–1001.
- Eeles, E.M.; White, S.V.; O’Mahony, S.M.; Bayer, A.J.; Hubbard, R.E. The impact of frailty and delirium on mortality in older inpatients. Age Ageing 2012, 41, 412–416.
- Strandberg, T.E.; Pitkälä, K.H. Frailty in elderly people. Lancet 2007, 369, 1328–1329.
- Fried, L.P.; Tangen, C.M.; Walston, J.; Newman, A.B.; Hirsch, C.; Gottdiener, J.; Seeman, T.; Tracy, R.; Kop, W.J.; Burke, G.; et al. Frailty in older adults: Evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, M146–M157.
- Rockwood, K.; Song, X.; MacKnight, C.; Bergman, H.; Hogan, D.B.; McDowell, I.; Mitnitski, A. A global clinical measure of fitness and frailty in elderly people. CMAJ 2005, 173, 489–495.
- Clegg, A. Frailty in elderly people. Lancet 2013, 381, 752–762.
- Song, X.; Mitnitski, A.; Rockwood, K. Prevalence and 10-year outcomes of frailty in older adults in relation to deficit accumulation. J. Am. Geriatr. Soc. 2010, 58, 681–687.
- Fried, L.P.; Ferrucci, L.; Darer, J.; Williamson, J.D.; Anderson, G. Untangling the concepts of disability, frailty, and comorbidity: Implications for improved targeting and care. J. Gerontol. 2004, 59, 255–263.
- Fried, L.P.; Ferrucci, L.; Darer, J.; Williamson, J.D.; Anderson, G. Phenotype of frailty: Characterization in the women’s health and aging studies. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 262–266.
- Morley, J.E.; Haren, M.T.; Rolland, Y.; Kim, M.J. Frailty. Med. Clin. N. Am. 2006, 90, 837–847.
- Flatt, T. A new definition of aging? Front. Genet. 2012, 3, 148.
- Giaimo, S.; d’Adda di Fagagna, F. Is cellular senescence an example of antagonistic pleiotropy? Aging Cell 2012, 11, 378–383.
- Sharpless, N.E. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91.
- Sager, R. Senescence as a mode of tumor suppression. Environ. Health Perspect. 1991, 93, 59–62.
- Baker, D.J. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236.
- Baker, D.J. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836.
- Baker, D.J. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435.
- Kaszubowska, L. Telomere shortening and ageing of the immune system. J. Physiol. Pharmacol. 2008, 59 (Suppl. S9), 169–186.
- Titus, S. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013, 5, 172ra121.
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263.
- Shimizu, I. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012, 15, 51–64.
- Minamino, T. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087.
- Ryan, A.S. Insulin resistance with aging: Effects of diet and exercise. Sports Med. 2000, 30, 327–346.
- Walters, M.S. Smoking accelerates aging of the small airway epithelium. Respir. Res. 2014, 15, 94.
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39.
- Xu, H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830.
- Roden, M. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865.
- Guo, N. Short telomeres compromise beta-cell signaling and survival. PLoS ONE 2011, 6, e17858.
- Yang, T.K. Davallialactone from mushroom reduced premature senescence and inflammation on glucose oxidative stress in human diploid fibroblast cells. J. Agric. Food Chem. 2013, 61, 7089–7095.
- Liu, J. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell. Signal. 2014, 26, 110–121.
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514.
- Kim, Y.J. miR-486–5p induces replicative senescence of human adipose tissue-derived mesenchymal stem cells and its expression is controlled by high glucose. Stem. Cells Dev. 2012, 21, 1749–1760.
- Salpea, K.D.; Humphries, S.E. Telomere length in atherosclerosis and diabetes. Atherosclerosis 2010, 209, 35–38.
- Kawashima, S.; Yokoyama, M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 998–1005.
- Barzilai, N.; Banerjee, S.; Hawkins, M.; Chen, W.; Rossetti, L. Caloric restriction reverses hepatic insulin resistance in aging rats by decreasing visceral fat. J. Clin. Investig. 1998, 101, 1353–1361.
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118.
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity May Accelerate the Aging Process. Front. Endocrinol. 2019, 10, 266.
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839.
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423.
- Bertrand, H.A.; Lynd, F.T.; Masoro, E.J.; Yu, B.P. Changes in adipose mass and cellularity through the adult life of rats fed ad libitum or a life-prolonging restricted diet. J. Gerontol. 1980, 35, 827–835.
- Liao, C.Y.; Rikke, B.A.; Johnson, T.E.; Diaz, V.; Nelson, J.F. Genetic variation in the murine lifespan response to dietary restriction: From life extension to life shortening. Aging Cell 2010, 9, 92–95.
- Speakman, J.R.; Mitchell, S.E. Caloric restriction. Mol. Asp. Med. 2011, 32, 159–221.
- Liao, C.Y. Fat maintenance is a predictor of the murine lifespan response to dietary restriction. Aging Cell 2011, 10, 629–639.
- Vermeij, W.P. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 2016, 537, 427–447.
- Huffman, K.M. Caloric restriction alters the metabolic response to a mixed-meal: Results from a randomized, controlled trial. PLoS ONE 2012, 7, e28190.
- Riera, C.E.; Dillin, A. Tipping the metabolic scales towards increased longevity in mammals. Nat. Cell Biol. 2015, 17, 196–203.
- Willette, A.A. Interleukin-8 and interleukin-10, brain volume and microstructure, and the influence of calorie restriction in old rhesus macaques. Age 2013, 35, 2215–2227.
- Youm, Y.H. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269.
- Yang, H.; Youm, Y.H.; Dixit, V.D. Inhibition of thymic adipogenesis by caloric restriction is coupled with reduction in age-related thymic involution. J. Immunol. 2009, 183, 3040–3052.
- Masoro, E.J. Overview of caloric restriction and ageing. Mech. Ageing Dev. 2005, 126, 913–922.
- Dillin, A. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401.
- Lee, S.S. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48.
- Trifunovic, A. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423.
- Kujoth, G.C. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484.
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Barja, G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27.
- Calabrese, E.J.; Bachmann, K.A.; Bailer, A.J.; Bolger, P.M.; Borak, J.; Cai, L.; Cedergreen, N.; Cherian, M.G.; Chiueh, C.C.; Clarkson, T.W.; et al. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007, 222, 122–128.
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766.
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711.
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 2014, 12, 288–341.
- López-Lluch, G. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773.
- Finley, L.W. Skeletal muscle transcriptional coactivator PGC-1α mediates mitochondrial, but not metabolic, changes during calorie restriction. Proc. Natl. Acad. Sci. USA 2012, 109, 2931–2936.
- Gomes, A.P. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638.
- Cuervo, A.M. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140.
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643.
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21.
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156.
- Maegawa, S. Caloric restriction delays age-related methylation drift. Nat. Commun. 2017, 8, 539.
- Redman, L.M. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 1–11.
- Willett, W.C.; Sacks, F.; Trichopoulou, A.; Drescher, G.; Ferro-Luzzi, A.; Helsing, E.; Trichopoulos, D. Mediterranean diet pyramid: A cultural model for healthy eating. Am. J. Clin. Nutr. 1995, 61 (Suppl. S6), S1402–S1406.
- Fundación Dieta Mediterránea. 2010. Available online: (accessed on 1 June 2019).
- Keys, A.B. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease; Harvard University Press: Cambridge, MA, USA, 1980; p. 381.
- Trichopoulou, A. Adherence to a Mediterranean Diet and Survival in a Greek Population. N. Engl. J. Med. 2003, 348, 2599–2608.
- Trichopoulou, A. Diet and overall survival in the elderly. BMJ 1995, 311, 1457–1460.
- Estruch, R. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet. N. Engl. J. Med. 2013, 368, 1279–1290.
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34.
- Sofi, F. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 337, a1344.
- Sofi, F. Accruing evidence on benefits of adherence to the Mediterranean diet on health: An updated systematic review and meta-analysis. Am. J. Clin. Nutr. 2010, 92, 1189–1196.
- Voelker, R. The Mediterranean Diet’s Fight against Frailty. JAMA 2018, 319, 1971–1972.
- Bach-Faig, A. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011, 14, 2274–2284.
- Kromhout, D. Comparative ecologic relationships of saturated fat, sucrose, food groups, and a Mediterranean food pattern score to 50-year coronary heart disease mortality rates among 16 cohorts of the Seven Countries Study. Eur. J. Clin. Nutr. 2018, 72, 1103–1110.


