 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kyoko Hida | + 7547 word(s) | 7547 | 2020-05-06 09:39:10 | | | |
| 2 | Kyoko Hida | Meta information modification | 7547 | 2020-05-11 15:51:11 | | | | |
| 3 | Rita Xu | -3864 word(s) | 3683 | 2020-05-12 05:05:41 | | | | |
| 4 | Rita Xu | -7 word(s) | 3676 | 2020-10-28 10:28:33 | | |
Video Upload Options
Tumor progression relies on angiogenesis from established normal vasculature to form new tumor blood vessels. Tumor endothelial cells (TECs) in the tumor blood vessels maintain tumor vessel formation through continual angiogenesis. TECs are heterogeneous with a diverse cellular origin. Moreover, the various factors and conditions in the tumor microenvironment elicit specific characteristics in TECs differentiating them from endothelial cells in normal vessels. TECs are the main focus of antiangiogenesis strategies, and their unique features make tumor blood vessels good anti-cancer therapeutic targets.
1. Tumor Vasculature
Tumors become vascularized through more than one mechanism of angiogenesis. It may take the form of sprouting angiogenesis [1] from preexisting vessels or the splitting of preexisting vessels into two daughter vessels by a process known as intussusception [2]. Neovascularization processes such as vasculogenesis mediated by endothelial progenitor cells (EPCs) recruited from the bone marrow can lead to the development of tumor blood vessels [3]. In addition, through the process of vasculogenic mimicry, highly invasive and metastatic melanoma cells mimic the endothelium-forming ability of endothelial cells (ECs) and create loops or networks resembling the vasculature, which are devoid of ECs but contain blood cells [4]. These channels facilitate tumor blood supply independent of angiogenesis. Breast, colon, lung, pancreatic, ovarian, glioblastoma multiforme, and hepatocellular carcinomas are among the cancer types that present with vasculogenic mimicry [5].
The tumor blood vessels carry nutrients to the tumor to stimulate rapid growth of the tumor, enrich the stroma with immune cells, and also aid tumor metastasis. In the wake of their development, tumors cause significant transformations in all cells and tissues in their surroundings. The growing tumor begins to exert physical pressure on the vessels, thus causing portions of the vessels to flatten and lose their lumen. Hierarchal vessel structure and blood flow are distorted (Figure 1A). Moreover, tumor-derived growth factors such as vascular endothelial growth factor (VEGF) stimulate rapid angiogenesis without sufficient control from angiogenesis inhibitors, which leads to the formation of tortuous vessels with loose EC junctions [6], little or no perivascular cell coverage [7], and an overall leaky nature, further contributing to the high interstitial fluid pressure observed in tumors [8][9].
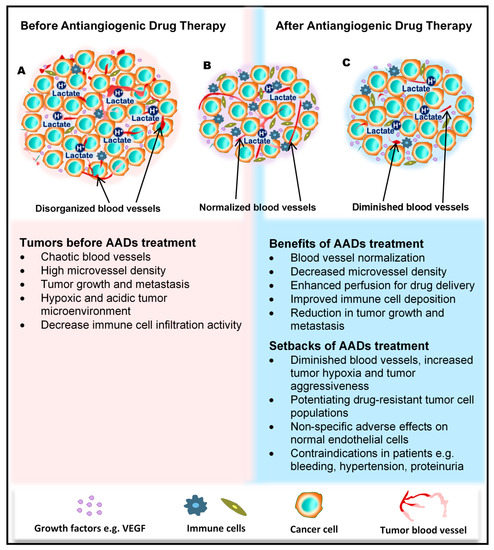
Figure 1. Benefits and side effects of antiangiogenic drugs. AADs, antiangiogenesis drugs. The dependency of tumors on their resident blood vessels to grow and metastasize has led to the targeting of tumor blood vessels to starve the tumor cells and close the metastasis portals. (A) Before the administration of AADs, the tumor histology is characterized by a high density of microvessels, with an undefined order of organization. The microenvironment is generally acidic, with high lactate levels, and immunologically suppressed. (B) However, after AAD therapy, tumor blood vessels become normalized, microvessel number reduces, tumor growth recedes, and immune cells infiltrate the tumors more through the normalized vasculature. (C) In addition to these benefits, AAD use causes some undesirable effects, including tumor hypoxia (from prolonged use of AADs) and destruction of normal vessels leading to bleeding. Patients may also experience hypertension and proteinuria, among others.
2. Angiogenesis and Its Inhibition in Tumors
2.1. Angiogenesis
Sprouting angiogenesis is the physiological process that was described as the formation of new blood vessels by capillary sprouting from preexisting vessels. Most blood vessels remain quiescent in the adult body, and angiogenesis occurs only in female reproductive organs and in the placenta during pregnancy. However, ECs preserve the function of rapid division in response to a physiological stimulus such as hypoxia or inflammation [10]. Angiogenic factors such as VEGF, basic fibroblast growth factor (bFGF), angiopoietin, and platelet-derived growth factor (PDGF) drive angiogenesis, and it is also performed as a normal process in growth and developmental processes, such as wound healing [11][12].
2.2. Factors That Stimulate and Regulate Tumor Angiogenesis
The tumor uses existing angiogenic mechanisms to induce capillary growth. Various growth factors, including VEGF, bFGF, PDGF, and angiopoietin, can induce tumor angiogenesis [13]. These factors are secreted from tumor cells and stromal cells. For example, tumors activate tumor-associated macrophages (TAMs) or neutrophils to produce angiogenic factors such as VEGF and matrix metalloproteinases (MMPs) [14][15]. Furthermore, other immune cell types indirectly influence the process of angiogenesis through the secretion of VEGF-A, bFGF, MMP9, interferon gamma (IFNγ), and interleukin-17 (IL-17) [16][17][18]. These angiogenesis stimuli may also be triggered by metabolic stress such as hypoxia, low pH or hypoglycemia, mechanical stress and genetic mutations, and p53 regulation [19][20][21][22].
2.3. Concept of Antiangiogenic Therapy and Development of Angiogenesis Drugs and Their Molecular Targets
For a long period of time, cytotoxic drugs were conventionally used for anticancer treatment. Later on, antiangiogenic therapy was proposed as a new concept for anticancer treatment. Dr. Judah Folkman in the early 1970s proposed that cancer could be treated by blocking the supply of oxygen and nutrients through the inhibition of tumor angiogenesis [23]. Moreover, antiangiogenic therapy has the potential to normalize blood vessel structures and improve systemic delivery of oxygen or perfusion of cytotoxic drugs into tumor tissues (Figure 1B) [24][25].
Bevacizumab, a monoclonal antibody targeting VEGF, was first approved as an antiangiogenic therapy in 2004 for the treatment of colon cancer in combination with chemotherapy [26][27]. Since then, various antiangiogenesis drugs, either as monotherapy or in combination with other cytotoxic drugs, have been developed, used in clinical trials, and approved for the treatment of cancer. The antiangiogenesis drugs include tyrosine kinase inhibitors (TKIs) such as sorafenib, sunitinib, axitinib, and pazopanib target receptors for VEGF and PDGF to inhibit the VEGF pathway [28]. By targeting their receptors expressed on tumor endothelial cells (TECs), these angiogenesis drugs are able to inhibit tumor angiogenesis.
Besides VEGF and its receptors, several other growth factors and receptors are involved in pathways that regulate tumor growth and angiogenesis in a complementary and coordinated manner. New multikinase inhibitors that can simultaneously target more than one of these pathways have been developed and approved for anticancer treatment. For example, regorafenib was found to inhibit a distinct set of receptor kinases, including the vascular endothelial growth factor receptors (VEGFR1–3), TIE2, fibroblast growth factor receptor 1 (FGFR1), and platelet-derived growth factor receptor beta (PDGFR-b), and has been approved for treating hepatocellular carcinoma, colorectal cancer, and gastrointestinal stromal tumors [29][30][31][32]. Cabozantinib was found to inhibit VEGFR2, c-Met, and AXL receptor tyrosine kinases and has been approved for treating metastatic renal cell carcinoma [33][34]. Several other multikinase inhibitors have also been developed and used in clinical trials or clinical settings [35]. The mammalian target of the rapamycin (mTOR) pathway is also involved in angiogenesis, and mTOR inhibitors such as everolimus and temsirolimus have been approved for clinical use [36].
2.4. Positive Achievements and Clinical Outcomes of Antiangiogenic Therapy
Antiangiogenic therapy enhances T-cell priming and activation by promoting dendritic cell maturation and increasing T-cell infiltration into the tumor tissue via tumor vessel normalization [37][38][39]. Antiangiogenic therapy also converts an immune-permissive tumor microenvironment by decreasing regulatory T-cell and myeloid-derived suppressor cells [39][40]. Tumor vessel normalization is crucial for improving the immune environment, tumor immunity is the key factor for anticancer treatment, and improving the immune environment is necessary to increase treatment efficacy (Figure 1B). Today, immune checkpoint inhibitors have been approved for treating various cancers [41][42][43]; furthermore, recent therapeutic strategies target both tumor angiogenesis and tumor immunity for achieving a greater therapeutic effect. As a combination therapy, bevacizumab and IFNα have been approved for treating metastatic renal cell carcinoma [44]. Clinical trials of combined TKIs and immune checkpoint inhibitors are actively being conducted, and favorable outcomes have been observed in metastatic renal cell carcinoma and non-small cell lung cancer [45][46][47][48].
2.5. Negative Side Effects/Adverse Responses from Patients
Regardless of the enormous benefits, antiangiogenic therapy has several problems. Complications such as hypertension, hand–foot syndrome, proteinuria, and thyroid dysfunction could occur as adverse events because of the effect of antiangiogenesis drugs on normal blood vessels [49]. Drug resistance to antiangiogenic therapy may also occur, and drug switching is generally required. Various mechanisms are described for explaining the resistance to antiangiogenic therapy, and various cellular and noncellular factors in the tumor microenvironment such as TECs, immune cells, cancer-associated fibroblasts, or extracellular matrix components are involved in these resistance mechanisms [50][51]. Long-term antiangiogenic therapy leads to tumor hypoxia and induces tumor aggressive behavior [52] (Figure 1C). It has been reported that hypoxia promotes the accumulation of TAMs [53] and induces tumor angiogenesis through the mobilization of bone marrow-derived endothelial precursor cells [54]. Hypoxia also induces chromosomal abnormalities in TECs via reactive oxygen species [55] and the selection of more invasive metastatic populations of tumor cells that are resistant to antiangiogenic therapy [56] (Figure 1C). Other modes of tumor vascularization such as vascular mimicry and vessel co-option have been suggested as another mechanism underlying the resistance to antiangiogenic therapy. Vascular mimicry is a tumor blood supply system without the participation of ECs. Vascular-like constructions are generated through the differentiation of tumor cells into endothelial-like cells, independent of conventional angiogenic factors [4]. An increase in vascular mimicry has been observed after antiangiogenic therapy [57]. Vessel co-option occurs in metastatic tumors. Tumor cells co-opt and grow around existing blood vessels [58]. Vessel co-option could explain the cause of resistance to antiangiogenic therapy in various cancers [59][60][61].
3. Tissue and Cellular Sources of TECs
3.1. Blood Vessels
The key players in the formation of blood vessels and the likely target of antiangiogenic therapy, ECs, through the process of sprouting angiogenesis, migrate and proliferate to form vessels by relying on cues from the growing tumor. Like the vessels recruited into the tumor, the ECs are similarly imparted by the growing tumor themselves as well as the microenvironmental factors, leading to the development of unique characteristics in these recruited ECs different from those of normal endothelial cells (NECs). These endothelial cells, often designated as TECs or tumor-associated ECs, have their primary origin as the blood vessels within the tumor mass. TECs have been isolated from various tumors, and analyses have shown that they indeed have an endothelial lineage [62]; however, some studies have demonstrated that other cellular sources of the tumor endothelium exist.
3.2. Cancer Stem Cells and EPCs
Cancer stem cells (CSCs) and EPCs are involved in this nonconventional tumor vasculature formation through vasculogenesis. ECs in glioblastoma were found to have similar genetic alterations as those in the tumor cells; moreover, glioblastoma stem cells positive for the stem cell marker CD133 were capable of generating cells that phenotypically and functionally resembled ECs [63][64]. Contrary to the above suggestions that TECs could arise from cancer stem cells, some recent reports have demonstrated that it is rather rare to find ECs with neoplastic origins within the glioblastoma vasculature. Kulla et al. reported that glomeruloid vessels microdissected from glioblastoma tissue specimens lacked mutations in the tumor protein p53 (TP53) gene as compared to the surrounding glioblastoma cells with a mutation in 3 exons of this gene [65]. Additionally, epidermal growth factor receptor (EGFR) gene amplification could not be identified in CD34+ endothelial cells within the vascular linings of glioblastoma tissue, further supporting the unlikely contribution of glioblastoma cells to tumor vessel formation [66]. However, glioma stem cells may contribute to tumor angiogenesis by differentiating into perivascular cells, like pericytes, not endothelial cells, which are necessary for blood vessel maturation [67] EPCs originate as progenitor cells from the bone marrow possessing the ability to develop into matured ECs. As EPCs travel from the bone marrow into the peripheral blood to the tumors, their surface molecules change from CD133+/CD34+/VEGFR2+ cells to cells with a decreased expression of CD133, while maintaining the expression of VEGFR2 and the hematopoietic progenitor cell antigen CD34 as they mature in circulation. The matured ECs in the vessels finally lose both CD133 and CD34 but display high VEGFR2 expression and are positive for vascular endothelial cadherin, von Willebrand factor, and platelet endothelial cell adhesion molecule 1 (PECAM1), also known as CD31 [68]. EPCs are known to both support vessel formation and integrate directly into the endothelium. Early-forming EPCs secrete angiogenic growth factors and cytokines (e.g., VEGF, stromal cell-derived factor-1 (SDF-1), insulin-like growth factor 1 (IGF-1), and IL-8 [69]) to facilitate neovascularization, whereas late-forming EPCs were found to be better at forming capillary tubes and could differentiate easily into ECs [70]. Both tumor-derived growth factors and EPC-secreted molecules play a role in recruiting EPCs into peripheral circulation and further into the tumor tissue to initiate tumor vasculogenesis. The abundance of EPCs in various types of cancers, including breast cancer [71], non-small cell lung cancer [72], hepatocellular carcinoma [73], colorectal cancer, leukemia, lymphoma, myeloma [74], and glioma [63][64], indicates their relevance in the tumor growth [75]. The role of EPCs in enhancing microvessel formation within some of these tumors has been demonstrated. Therefore, EPCs are an established source of TECs.
4. TEC Characteristics
4.1. Cytogenetic Abnormality
Contrary to earlier assumptions about TECs, it has been shown for over a decade that TECs are undoubtedly different from NECs. TECs have characteristics that are considered to be abnormal, ranging from their morphology to genetics and function (Figure 2). TECs obtained from human melanoma and liposarcoma tumor xenografts structurally possessed bigger nuclei with different size variations than those in NECs [76]. These nuclei were made up of chromosomes with various structural aberrations, translocations, chromosomal aneuploidy, missing chromosomes, and the presence of additional chromosomes such as double minutes and some of unknown origin [76]. Microvascular ECs in B-cell lymphoma were also found to have lympho-specific chromosomal translocations [77]. More recently, nonhematopoietic aneuploid CD31+ circulating TECs were detected in the peripheral blood of patients with breast cancer, demonstrating that circulating TECs, and not only tumor-bound TECs, possess chromosomal changes [78]. Due to these changes, TECs obtained from human [79] and murine tumors [76] are genetically unstable compared with NECs. We have reported that the TECs isolated from xenograft human epithelial tumors, which were CD133(+), were susceptible to a higher frequency of aneuploidy than the CD133(-) TECs. This suggests that progenitor cells do not only contribute to TEC generation but may also be involved in inducing genetic instability in these cells [79]. Other causes of TEC aneuploidy include hypoxia-induced reactive oxygen species and VEGF signaling [55]. It has been reported that VEGF signaling also regulates the centrosome duplication cycle in ECs and induces centrosome overduplication [80], which could further lead to aneuploidy [81]. Stromal cells like fibroblasts and ECs receive tumor genetic material (DNA/chromosomes) transferred via apoptotic bodies from tumor cells [82][83]. Such horizontal gene transfer can lead to cytogenetic alterations in ECs. Furthermore, genetic instabilities could occur within the cells through the propagation of the DNA. Replication of the transferred DNA may occur in the recipient cells provided they have undergone certain changes including p21 and p53 inactivation [82][83][84].
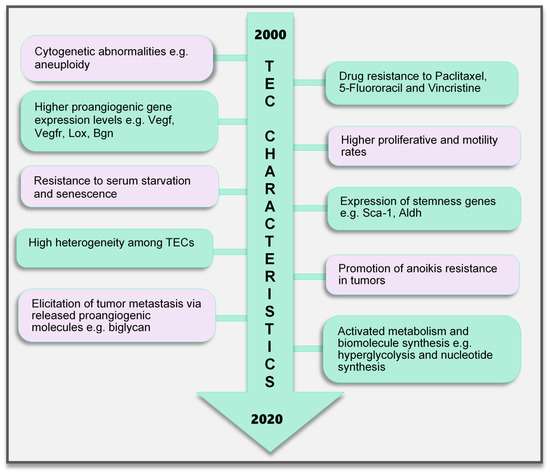
Figure 2. TEC characteristics identified to date. Various characteristics of TECs have been observed, which make them unique when compared with endothelial cells in normal blood vessels. These range from the genetic composition and expression of genes, abnormal karyotype, higher biological activities (proliferation and motility), and TEC influence on tumor cells to modulate cancer cell metastasis and survival to modifications in their metabolic signature. TECs are not normal and their abnormality creates a targetable avenue to influence the growth of tumors and improve therapeutics in cancer treatment. TEC, tumor endothelial cells
4.2. Genotypic Changes
In addition, genes regulating angiogenesis, cell proliferation and motility, stemness, and drug resistance, among others, are altered in TECs. ECs require the autocrine function of VEGF to sustain vascular integrity and viability [85]. VEGF acts through its receptors, the VEGFRs. Among them, VEGFR1 and VEGFR2 have been shown to be highly expressed in TECs compared to those in NECs. This enhances the response of TECs to VEGF more than NECs [86], which makes TECs proangiogenic and may also support their ability to survive in serum-free media unlike their normal counterparts [87]. Furthermore, TECs proliferate and migrate faster than NECs [86]. A study comparing ECs isolated from colorectal cancer and normal colorectal mucosa demonstrated the unique expression of 46 genes in the tumor endothelium (i.e., TECs) as opposed to the normal endothelium [88]. The top 25 genes included MMP2 and MMP11, as well as variations of collagen types I, III, and VI, among others. The authors of that study described these genes as tumor endothelial markers (TEMs) by confirming through the analysis of TEM7 expression in the lung, pancreas, breast, and brain and suggested that the TEMs may be expressed in other cancers as well [88]. These findings established a promising future for the use of the TEMs in further research to identify novel antiangiogenesis strategies. However, later publications suggested that not all the TEMs are unique to TECs. TEM1, TEM 5, TEM7, and TEM 8 were found to be also expressed in normal cells, tissues, and organs [89][90][91][92]. The secreted and membrane forms of TEM7 were observed in various osteogenic sarcoma cell lines [93][95]. TEC heterogeneity is a major factor contributing to the genotypic differences observed between NECs and TECs. TECs obtained from low-metastatic and high-metastatic tumors have different tendencies toward cell proliferation, motility, and drug resistance, indicating that there may be differences in the underlying genotype of these cells. We had demonstrated that genes required for angiogenesis-related molecules and matrix-degrading enzymes were upregulated in the TECs obtained from highly metastatic tumors [94]. Moreover, the heterogeneity arising from the different cellular origin (stems cells, cancers, and EPCs) of TECs may account for the different genotype of TECs compared with NECs [95]. TECs isolated from different tumor types show variations in their gene expression profiles, the upregulated gene, or the gene set classification. ADAM23, FAP, GPNMB, and PRSS3 genes were high in ovarian cancer ECs [96], while G-protein-coupled receptor RDC1 was the distinctively induced TEM in TECs of the brain and the peripheral vasculature [97]. Furthermore, fewer similarities exist between different TEC gene profiles. A collation of 73 TEC marker genes from five studies showed that at most, only two cancer types shared one of these five genes (EGFL6, HEYL, MMP9, SPARC, PLXDC1), and the rest were expressed uniquely in only one cancer [98]. Aird reviewed that TEC gene expression heterogeneity is influenced by the tumor type, extracellular environment, and epigenetic regulation [98]; however, the current vast database needs to be sifted through by further research to obtain significant translational benefits.
4.3. Drug Resistance and Anoikis Resistance
A drug resistance phenotype has been reported in various TECs. The TECs isolated from A375SM (super-metastatic human melanoma cells) xenografts are resistant to the anticancer drug paclitaxel [99], and CD105+ TECs isolated from hepatocellular carcinoma were found to be resistant to doxorubicin and 5-fluorouracil [100]. TEC resistance to paclitaxel was attributed to the upregulated expression of multidrug resistance protein 1 (MDR1) mRNA through VEGF signaling [99]. The ATP-binding cassette (ABC) transporter superfamily, which includes ABCB1 (i.e., MDR1/P-glycoprotein, P-gp), is expressed in CSCs [101]. TECs may possess stemness properties as they express MDR1 and other stemness markers such as aldehyde dehydrogenase (ALDH), CD90, and stem cell antigen-1 (Sca-1) [102]. These stem cell features of TECs suggest the versatility in their function and continuous availability.
The survival and function of TECs in the tumor microenvironment can be promoted by the expression of microRNAs (miR-145), which confers on the cells the ability to resist anoikis and become more adhesive by activating ERK1/2 and epigenetic modifications to enhance miR-145 expression [103]. Circulating tumor-associated ECs that express Bcl-2 have been implicated in protecting tumor cells from anoikis and enhancing lung metastasis. For TECs to protect tumor cells in circulation, they should also possess the ability to survive in circulation [104]. Although the study did not address anoikis resistance in TECs, the authors showed that TECs exhibited increased adhesive properties and were Bcl-2-positive, which was similar to the effect of miR145 on anoikis-resistant TECs in our study. Bcl-2 upregulation has been shown to enhance EC survival [105]. These findings may imply that anoikis-resistant TECs could promote tumor metastasis. In fact, we have demonstrated that TECs can elicit metastasis of low-metastatic tumors to the lung by releasing the angiocrine factor biglycan [106].
4.4. Altered Metabolism
EC metabolism plays a crucial role in the formation of blood vessels. In general, most of the known metabolic–biosynthetic pathways are more activated in TECs than in NECs [107]. Glycolysis regulates the migration and proliferation of tip and stalk ECs, respectively, through the activity of the glycolytic enzyme phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) [108]. Although normal healthy ECs were shown to have a higher glycolytic level than other healthy cells [108], we and other groups have demonstrated that glycolysis is more activated in TECs [109], conferring on these cells the “hyperglycolytic” label [110][111]. In these hyperglycolytic TECs, PFKFB3 and other glycolytic enzymes are upregulated compared to those in their normal counterparts. Inhibition of PFKFB3 in ECs decreased tumor metastasis by inducing the normalization of the tumor vessels. The vessel structure was improved from a disorganized structure to more matured pericyte-covered, perfused vessels, which supported better drug delivery leading to antimetastatic therapeutic benefits [110]. In addition, cyclooxygenase 2 (COX2) supports the upregulated expression of PFKFB3 and VEGF in TECs to facilitate the higher glycolytic rates in these cells [111]. This role of COX2 in TEC glycolysis may partly explain our previous discovery that COX2 is essential for tumor angiogenesis. We found that COX2 was upregulated in TECs and its inhibition decreased the tumor growth and the recruitment of vascular progenitor cells into tumor vessels. COX2 inhibition further reduced the migration and proliferation of TECs, implying that both the angiogenic activity of resident TECs in the tumor and the recruitment of progenitor cells that can become TECs are impaired by COX2 [112]. The hyperglycolytic nature of TECs may potentially contribute to the overwhelming lactate burden within the tumor and exert various effects on stromal cells. It is interesting to note that somehow TECs survive in these high-lactate environments, and angiogenesis is not impaired. We have reported that in addition to the enzymes that directly influence substrate metabolism, enzymes that maintain intracellular pH at physiological levels significantly influence TEC proliferation. TECs release more lactate extracellularly than NECs, accompanied by an acidification of the culture media. Notably, these TECs displayed an upregulated expression of the pH regulatory enzyme carbonic anhydrase 2 (CAII). CAII in TECs was required for successful proliferation, independent of the metabolic substrate available [109]. Moreover, to maintain their higher proliferative rates, TECs require nucleotide precursors and lipids from serine and lipid biosynthesis pathways, respectively. To support this demand for biomolecules, TECs express higher levels of key pathway enzymes such as D-3-phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase 1 (PSAT1) [110] for serine biosynthesis and fatty acid synthase (FASN) [113] for lipid synthesis. Nucleotide biosynthesis is also enhanced in TECs compared to that in NECs [107]. TECs are a robust group of cells that deserve special attention and study in the pursuit of direct, effective tumor angiogenesis therapeutic strategies.
References
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J; Endothelial Function and Dysfunction. Testing and Clinical Relevance. Circulation 2007, 115, 1285–1295.
- Zecchin, A.; Borgers, G.; Carmeliet, P. Endothelial cells and cancer cells: Metabolic partners in crime? Curr. Opin. Hematol. 2015, 22, 234–242.
- Robert F. Furchgott; John V. Zawadzki; The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373-376, 10.1038/288373a0.
- Ryszard J. Gryglewski; Stuart Bunting; Salvador Moncada; Roderick J. Flower; John R. Vane; Arterial walls are protected against deposition of platelet thrombi by a substance (prostaglandin X) which they make from prostaglandin endoperoxides. Prostaglandins 1976, 12, 685-713, 10.1016/0090-6980(76)90047-2.
- D. Leung; G Cachianes; W. Kuang; D. Goeddel; N Ferrara; Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306-1309, 10.1126/science.2479986.
- Dudley, A.C; Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536.
- Lin Xiao; Dae Joong Kim; Clayton L. Davis; James V. McCann; James M. Dunleavey; Alissa K. Vanderlinden; Nuo Xu; Samantha G. Pattenden; Stephen V. Frye; Xia Xu; et al.Mark OnaitisElizabeth Monaghan-BensonKeith BurridgeAndrew C. Dudley Tumor Endothelial Cells with Distinct Patterns of TGFβ-Driven Endothelial-to-Mesenchymal Transition. Cancer Research 2015, 75, 1244-54, 10.1158/0008-5472.CAN-14-1616.
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschläger, M.; et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 2019.
- Takashi Aizawa; Hideaki Karasawa; Ryo Funayama; Matsuyuki Shirota; Takashi Suzuki; Shimpei Maeda; Hideyuki Suzuki; Akihiro Yamamura; Takeshi Naitoh; Keiko Nakayama; Michiaki Unno; Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer.. Cancer Medicine 2019, 8, 6370-6382, 10.1002/cam4.2523.
- N Kramer; J Schmöllerl; C Unger; H Nivarthi; A Rudisch; D Unterleuthner; M Scherzer; A Riedl; M Artaker; I Crncec; D Lenhardt; T Schwarz; B Prieler; Xiaonan Han; M Hengstschläger; J Schüler; Robert Eferl; Richard H Moriggl; W Sommergruber; Helmut Dolznig; Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene 2017, 36, 5460-5472, 10.1038/onc.2017.144.
- Erik Sahai; Igor Astsaturov; Edna Cukierman; David G. DeNardo; Mikala Egeblad; Ronald M. Evans; Douglas Fearon; Florian R. Greten; Sunil R. Hingorani; Tony Hunter; Richard O. Hynes; Rakesh K. Jain; Tobias Janowitz; Claus Jorgensen; Alec C. Kimmelman; Mikhail G. Kolonin; Robert G. Maki; R. Scott Powers; Ellen Puré; Daniel C. Ramirez; Ruth Scherz-Shouval; Mara H. Sherman; Sheila Stewart; Thea D. Tlsty; David A. Tuveson; Fiona M. Watt; Valerie Weaver; Ashani T. Weeraratna; Zena Werb; A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer 2020, 20, 174-186, 10.1038/s41568-019-0238-1.
- Xueman Chen; Erwei Song; Turning foes to friends: targeting cancer-associated fibroblasts. Nature Reviews Drug Discovery 2018, 18, 9-115, 10.1038/s41573-018-0004-1.
- Michel Félétou; The Endothelium, Part I: Multiple Functions of the Endothelial Cells -- Focus on Endothelium-Derived Vasoactive Mediators. Colloquium Series on Integrated Systems Physiology: From Molecule to Function 2011, 3, 1-306, 10.4199/c00031ed1v01y201105isp019.
- Kyoko Hida; Nako Maishi; Chisaho Torii; Yasuhiro Hida; Tumor angiogenesis—characteristics of tumor endothelial cells. International Journal of Clinical Oncology 2016, 21, 206-212, 10.1007/s10147-016-0957-1.
- Francesco De Sanctis; Stefano Ugel; John Facciponte; Andrea Facciabene; The dark side of tumor-associated endothelial cells. Seminars in Immunology 2018, 35, 35-47, 10.1016/j.smim.2018.02.002.
- Hida, K.; Maishi, N; Abnormalities of tumor endothelial cells and cancer progression. Oral Sci. Int. 2018, 15, 1-6.
- Kohei Matsuda; Noritaka Ohga; Yasuhiro Hida; Chikara Muraki; Kunihiko Tsuchiya; Takuro Kurosu; Tomoshige Akino; Shou-Ching Shih; Yasunori Totsuka; Michael Klagsbrun; Masanobu Shindoh; Kyoko Hida; Isolated tumor endothelial cells maintain specific character during long-term culture. Biochemical and Biophysical Research Communications 2010, 394, 947-954, 10.1016/j.bbrc.2010.03.089.
- Hitomi Ohmura-Kakutani; Kosuke Akiyama; Nako Maishi; Noritaka Ohga; Yasuhiro Hida; Taisuke Kawamoto; Junichiro Iida; Masanobu Shindoh; Kunihiko Tsuchiya; Nobuo Shinohara; Kyoko Hida; Identification of Tumor Endothelial Cells with High Aldehyde Dehydrogenase Activity and a Highly Angiogenic Phenotype. PLOS ONE 2014, 9, e113910, 10.1371/journal.pone.0113910.
- Valentina Fonsato; Stefano Buttiglieri; Maria Chiara Deregibus; Valeria Puntorieri; Benedetta Bussolati; Giovanni Camussi; Expression of Pax2 in Human Renal Tumor-Derived Endothelial Cells Sustains Apoptosis Resistance and Angiogenesis. The American Journal of Pathology 2006, 168, 706-713, 10.2353/ajpath.2006.050776.
- K. Hida; Tumor-Associated Endothelial Cells with Cytogenetic Abnormalities. Cancer Research 2004, 64, 8249-8255, 10.1158/0008-5472.can-04-1567.
- Tomoshige Akino; Kyoko Hida; Yasuhiro Hida; Kunihiko Tsuchiya; Deborah Freedman; Chikara Muraki; Noritaka Ohga; Kouhei Matsuda; Kousuke Akiyama; Toru Harabayashi; et al.Nobuo ShinoharaKatsuya NonomuraMichael KlagsbrunMasanobu Shindoh Cytogenetic Abnormalities of Tumor-Associated Endothelial Cells in Human Malignant Tumors. The American Journal of Pathology 2009, 175, 2657-2667, 10.2353/ajpath.2009.090202.
- Kyoko Hida; Nako Maishi; Dorcas A. Annan; Yasuhiro Hida; Contribution of Tumor Endothelial Cells in Cancer Progression. International Journal of Molecular Sciences 2018, 19, 1272, 10.3390/ijms19051272.
- Hisamichi Naito; Taku Wakabayashi; Hiroyasu Kidoya; Fumitaka Muramatsu; Kazuhiro Takara; Daisuke Eino; Keitaro Yamane; Tomohiro Iba; Nobuyuki Takakura; Endothelial side population cells contribute to tumor angiogenesis and anti-angiogenic drug resistance. Cancer Research 2016, 76, 3200-3210, 10.1158/0008-5472.can-15-2998.
- Aird, W.C.; Phenotypic Heterogeneity of the Endothelium. Circ. Res. 2007, 100, 158–173, 10.1161/01.res.0000255690.03436.ae.
- Yong S. Chang; Emmanuelle Di Tomaso; Nald M. McDonald; Rosemary Jones; Rakesh K. Jain; L.L. Munn; Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proceedings of the National Academy of Sciences 2000, 97, 14608-14613, 10.1073/pnas.97.26.14608.
- Hiroya Hashizume; Peter Baluk; Shunichi Morikawa; John W. McLean; Gavin Thurston; Sylvie Roberge; Rakesh K. Jain; Donald M. McDonald; Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. The American Journal of Pathology 2000, 156, 1363-1380, 10.1016/s0002-9440(10)65006-7.
- J. Denekamp; B. Hobson; Endothelial-cell proliferation in experimental tumours.. British Journal of Cancer 1982, 46, 711-720.
- Nako Maishi; Hiroshi Kikuchi; Masumi Sato; Hiroko Nagao-Kitamoto; Dorcas A. Annan; Shogo Baba; Takayuki Hojo; Misa Yanagiya; Yusuke Ohba; Genichiro Ishii; Kenkichi Masutomi; Nobuo Shinohara; Yasuhiro Hida; Kyoko Hida; Development of Immortalized Human Tumor Endothelial Cells from Renal Cancer. International Journal of Molecular Sciences 2019, 20, 4595, 10.3390/ijms20184595.
- Dhara N. Amin; Kyoko Hida; Diane R Bielenberg; Michael Klagsbrun; Tumor Endothelial Cells Express Epidermal Growth Factor Receptor (EGFR) but not ErbB3 and Are Responsive to EGF and to EGFR Kinase Inhibitors. Cancer Research 2006, 66, 2173-2180, 10.1158/0008-5472.can-05-3387.
- Yasufumi Sato; Persistent vascular normalization as an alternative goal of anti?angiogenic cancer therapy. Cancer Science 2011, 102, 1253-1256, 10.1111/j.1349-7006.2011.01929.x.
- Helfrich, I.; Scheffrahn, I.; Bartling, S.; Weis, J.; Felbert, V.V.; Middleton, M.; Kato, M.; Ergün, S.; Augustin, H.G.; Schadendorf, D; et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 2010, 207, 491–503.
- Sarah M. Taylor; Kathleen R. Nevis; Hannah L. Park; Gregory C. Rogers; Stephen L. Rogers; Jean Cook; Victoria L. Bautch; Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood 2010, 116, 3108-3117, 10.1182/blood-2010-01-266197.
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al.et al Heterogeneity of Tumor Endothelial Cells. Am. J. Pathol. 2012, 180, 1294–1307.
- Lyssiotis, C.A.; Kimmelman, A.C; Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875.
- Annalisa Zecchin; Joanna Kalucka; Charlotte Dubois; Peter Carmeliet; How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Frontiers in Immunology 2017, 8, 1750, 10.3389/fimmu.2017.01750.
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al.et al Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968-985, 10.1016/j.ccell.2016.10.006.
- Annan, D.A.; Maishi, N.; Soga, T.; Dawood, R.; Li, C.; Kikuchi, H.; Hojo, T.; Morimoto, M.; Kitamura, T.; Alam, M.T.; et al.et al Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Communication and Signaling 2019, 17, 169, 10.1186/s12964-019-0478-4.
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al.et al Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651-663, 10.1016/j.cell.2013.06.037.
- Longhou Fang; Soo-Ho Choi; Ji Sun Baek; Chao Liu; Felicidad Almazan; Florian Ulrich; Philipp Wiesner; Adam Taleb; Elena Deer; Jennifer Pattison; Jesús Torres-Vázquez; Andrew C. Li; Yury I. Miller; Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118-122, 10.1038/nature12166.
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S; Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 2009, 23, 3865–3873.
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al.et al Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197.
- Kimberly Krautkramer; Julia H. Kreznar; Kymberleigh A. Romano; Eugenio I. Vivas; Gregory A. Barrett-Wilt; Mary E. Rabaglia; Mark P. Keller; Alan D. Attie; Federico E Rey; John M. Denu; et al. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Molecular Cell 2016, 64, 982-992, 10.1016/j.molcel.2016.10.025.
- Anaïs Alves; Arthur Bassot; Anne-Laure Bulteau; Luciano Pirola; Béatrice Morio; Glycine Metabolism and Its Alterations in Obesity and Metabolic Diseases. Nutrients 2019, 11, 1356, 10.3390/nu11061356.
- Loscalzo, J.; Handy, D.E. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease (2013 Grover Conference Series). Pulm. Circ. 2014, 4, 169–174.
- Cathérine Dupont; D. Randall Armant; C. A. Brenner; Epigenetics: definition, mechanisms and clinical perspective.. Seminars in Reproductive Medicine 2009, 27, 351-357, 10.1055/s-0029-1237423.
- Luciano Pirola; Oskar Ciesielski; Aneta Balcerczyk; The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting. Cancers 2018, 10, 268, 10.3390/cancers10080268.
- Søreide, K. Cancer Epigenetics. In Handbook of Epigenetics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 519–534.
- Aditi Mehta; Stephanie Dobersch; Addi J. Romero-Olmedo; Guillermo Barreto; Epigenetics in lung cancer diagnosis and therapy. Cancer and Metastasis Reviews 2015, 34, 229-241, 10.1007/s10555-015-9563-3.
- Ewa Michalak; Marian L. Burr; Andrew J. Bannister; Mark A. Dawson; The roles of DNA, RNA and histone methylation in ageing and cancer. Nature Reviews Molecular Cell Biology 2019, 20, 573-589, 10.1038/s41580-019-0143-1.
- A. E. Morgan; T. J. Davies; Mark T. Mc Auley; The role of DNA methylation in ageing and cancer. Proceedings of the Nutrition Society 2018, 77, 412-422, 10.1017/s0029665118000150.
- Siavash K. Kurdistani; Histone Modifications in Cancer Biology and Prognosis. Epigenetics and Disease 2010, 67, 91-106, 10.1007/978-3-7643-8989-5_5.
- Yan, M.S.; Marsden, P.A; Epigenetics in the Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2297–2306.
- Majerski, A.A.; Quinton, A.C.; Marsden, P.A. Epigenetic Mechanisms of the Vascular Endothelium. Epigenet. Epigenom. 2014.
- Jessilyn Dunn; Salim Thabet; Hanjoong Jo; Flow-Dependent Epigenetic DNA Methylation in Endothelial Gene Expression and Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 2015, 35, 1562-1569, 10.1161/ATVBAHA.115.305042.
- Jason E. Fish; Charles C. Matouk; Alisa Rachlis; Steve Lin; Sharon C. Tai; Cheryl D'abreo; Philip A. Marsden; The Expression of Endothelial Nitric-oxide Synthase Is Controlled by a Cell-specific Histone Code. Journal of Biological Chemistry 2005, 280, 24824-24838, 10.1074/jbc.m502115200.
- Sarah Costantino; Epigenetic mechanisms of vascular dysfunction in obesity and type 2 diabetes. Cardiovascular Medicine 2019, 22, w02066, 10.4414/cvm.2019.02064.
- Ding-Yu Lee; Jeng-Jiann Chiu; Atherosclerosis and flow: roles of epigenetic modulation in vascular endothelium.. Journal of Biomedical Science 2019, 26, 56, 10.1186/s12929-019-0551-8.
- Peter A. Jones; Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Microbiology 2012, 13, 484-492, 10.1038/nrg3230.
- Chan, Y.; Fish, J.E.; Dabreo, C.; Lin, S.; Robb, G.B.; Teichert, A.-M.; Karantzoulis-Fegaras, F.; Keightley, A.; Steer, B.M.; Marsden, P.A; et al. The Cell-specific Expression of Endothelial Nitric-oxide Synthase. J. Biol. Chem. 2004, 279, 35087–35100.
- Shirodkar, A.V.; Bernard, R.S.; Gavryushova, A.; Kop, A.; Knight, B.J.; Yan, M.S.-C.; Man, H.-S.J.; Sud, M.; Hebbel, R.P.; Oettgen, P.; et al. A mechanistic role for DNA methylation in endothelial cell (EC)-enriched gene expression: Relationship with DNA replication timing. Blood 2013, 121, 3531–3540.
- Debby M.E.I. Hellebrekers; Veerle Melotte; Emmanuelle Viré; Elise Langenkamp; Grietje Molema; François Fuks; James G. Herman; Wim Van Criekinge; Arjan W. Griffioen; Manon Van Engeland; Identification of Epigenetically Silenced Genes in Tumor Endothelial Cells. Cancer Research 2007, 67, 4138-4148, 10.1158/0008-5472.can-06-3032.
- Debby M.E.I. Hellebrekers; Karolien Castermans; Ruud Dings; Nicole T.H. Hoebers; Kevin H. Mayo; Mirjam G. A. Oude Egbrink; Grietje Molema; François Fuks; Manon Van Engeland; Arjan W. Griffioen; Emmanuelle Viré; Epigenetic Regulation of Tumor Endothelial Cell Anergy: Silencing of Intercellular Adhesion Molecule-1 by Histone Modifications. Cancer Research 2006, 66, 10770-10777, 10.1158/0008-5472.can-06-1609.
- Wei Luo; Qiang Hu; Dan Wang; Kristin K. Deeb; Yingyu Ma; Carl D. Morrison; Song Liu; Candace S. Johnson; Donald L. Trump; Isolation and genome-wide expression and methylation characterization of CD31+ cells from normal and malignant human prostate tissue. Oncotarget 2013, 4, 1472-1483, 10.18632/oncotarget.1269.
- Kristin K. Deeb; Wei Luo; Adam R. Karpf; Angela R. Omilian; Wiam Bshara; Lili Tian; Michael A. Tangrea; Carl D. Morrison; Candace S. Johnson; Donald L. Trump; Differential vitamin D 24-hydroxylase/CYP24A1 gene promoter methylation in endothelium from benign and malignant human prostate. Epigenetics 2011, 6, 994-1000, 10.4161/epi.6.8.16536.
- B. St. Croix; Joel Bourne; Genes Expressed in Human Tumor Endothelium. Science 2000, 289, 1197-1202, 10.1126/science.289.5482.1197.
- Dylan T. Jones; Tanguy Lechertier; Richard Mitter; John M. J. Herbert; Roy Bicknell; J. Louise Jones; Ji-Liang Li; Francesca M. Buffa; Adrian L. Harris; Kairbaan Hodivala-Dilke; Gene Expression Analysis in Human Breast Cancer Associated Blood Vessels. PLOS ONE 2012, 7, e44294, 10.1371/journal.pone.0044294.
- Ivy Chung; Adam R. Karpf; Josephia R. Muindi; Jeffrey M. Conroy; Norma J. Nowak; Candace S. Johnson; Nald L. Trump; Epigenetic Silencing of CYP24 in Tumor-derived Endothelial Cells Contributes to Selective Growth Inhibition by Calcitriol. Journal of Biological Chemistry 2007, 282, 8704-8714, 10.1074/jbc.m608894200.
- Bin Liu; Tonghong Xu; Xinning Xu; Yuzhu Cui; Xiaojing Xing; Biglycan promotes the chemotherapy resistance of colon cancer by activating NF-κB signal transduction. Molecular and Cellular Biochemistry 2018, 449, 285-294, 10.1007/s11010-018-3365-1.
- Nako Maishi; Yusuke Ohba; Kosuke Akiyama; Noritaka Ohga; Jun-Ichi Hamada; Hiroko Nagao-Kitamoto; Mohammad Towfik Alam; Kazuyuki Yamamoto; Taisuke Kawamoto; Nobuo Inoue; Akinobu Taketomi; Masanobu Shindoh; Yasuhiro Hida; Kyoko Hida; Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Scientific Reports 2016, 6, 28039, 10.1038/srep28039.
- Andrew J. Bannister; Tony Kouzarides; Regulation of chromatin by histone modifications. Cell Research 2011, 21, 381-395, 10.1038/cr.2011.22.
- Anton Eberharter; Peter B Becker; Histone acetylation: a switch between repressive and permissive chromatin. EMBO reports 2002, 3, 224-229, 10.1093/embo-reports/kvf053.
- Philip Gregory; Klaus Wagner; Wolfram Hörz; Histone Acetylation and Chromatin Remodeling. Experimental Cell Research 2001, 265, 195-202, 10.1006/excr.2001.5187.
- Andrew J. Bannister; Robert Schneider; Tony Kouzarides; Histone Methylation. Cell 2002, 109, 801-806, 10.1016/s0092-8674(02)00798-5.
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays 2016, 39, 1–12.
- Wei Chen; Methode Bacanamwo; David G. Harrison; Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. Journal of Biological Chemistry 2008, 283, 16293-16298, 10.1074/jbc.M801803200.
- Carmen Urbich; Lothar Rössig; David Kaluza; Michael Potente; Jes-Niels Boeckel; Andrea Knau; Florian Diehl; Jian-Guo Geng; Wolf-Karsten Hofmann; Andreas M. Zeiher; Stefanie Dimmeler; HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 2009, 113, 5669-5679, 10.1182/blood-2009-01-196485.
- Deokbum Park; Hyunmi Park; Youngmi Kim; Hyuna Kim; Dooil Jeoung; HDAC3 acts as a negative regulator of angiogenesis. BMB Reports 2014, 47, 227–232.
- Roshana Thambyrajah; Muhammad Z.H. Fadlullah; Martin Proffitt; Rahima Patel; Shaun Cowley; Valerie Kouskoff; Georges Lacaud; HDAC1 and HDAC2 Modulate TGF-β Signaling during Endothelial-to-Hematopoietic Transition. Stem Cell Reports 2018, 10, 1369-1383, 10.1016/j.stemcr.2018.03.011.
- Andriana Margariti; Anna Zampetaki; Qingzhong Xiao; Boda Zhou; Eirini Karamariti; Daniel Martin; Xiaoke Yin; Manuel Mayr; Hongling Li; Zhongyi Zhang; Elena De Falco; Yanhua Hu; Gillian Cockerill; Qingbo Xu; Lingfang Zeng; Histone Deacetylase 7 Controls Endothelial Cell Growth Through Modulation of -Catenin. Circulation Research 2010, 106, 1202-1211, 10.1161/circresaha.109.213165.
- Madalena Barroso; Derrick Kao; Henk J. Blom; Isabel Tavares De Almeida; Maria Rita Azevedo E Castro; Joseph Loscalzo; Diane Handy; S-adenosylhomocysteine induces inflammation through NFkB: A possible role for EZH2 in endothelial cell activation.. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2015, 1862, 82-92, 10.1016/j.bbadis.2015.10.019.
- Aneta Balcerczyk; Dorota Rybaczek; Martyna Wojtala; Luciano Pirola; Jun Okabe; Assam El-Osta; Pharmacological inhibition of arginine and lysine methyltransferases induces nuclear abnormalities and suppresses angiogenesis in human endothelial cells. Biochemical Pharmacology 2016, 121, 18-32, 10.1016/j.bcp.2016.09.013.
- Martyna Wojtala; Ewa Macierzyńska-Piotrowska; Dorota Rybaczek; Luciano Pirola; Aneta Balcerczyk; Pharmacological and transcriptional inhibition of the G9a histone methyltransferase suppresses proliferation and modulates redox homeostasis in human microvascular endothelial cells. Pharmacological Research 2018, 128, 252-263, 10.1016/j.phrs.2017.10.014.
- Yang Duan; Xue Wu; Qiang Zhao; Jie Gao; Dawei Huo; Xinhua Liu; Zheng Ye; Xu Dong; Zheng Fu; Yongfeng Shang; Chenghao Xuan; DOT1L promotes angiogenesis through cooperative regulation of VEGFR2 with ETS-1. Oncotarget 2016, 7, 69674-69687, 10.18632/oncotarget.11939.
- Hai-Na Zhang; Qiao Xu; Abhimanyu Thakur; Martin Omondi Alfred; Manas Chakraborty; Arunima Ghosh; Xu-Ben Yu; Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sciences 2018, 213, 258-268, 10.1016/j.lfs.2018.10.028.
- Francesca Orso; Lorena Quirico; Daniela Dettori; Roberto Coppo; Federico Virga; Livia C Ferreira; Camilla Paoletti; Désirée Baruffaldi; Elisa Penna; Daniela Taverna; Role of miRNAs in tumor and endothelial cell interactions during tumor progression. Seminars in Cancer Biology 2020, 60, 214-224, 10.1016/j.semcancer.2019.07.024.
- Shusheng Wang; Arin B. Aurora; Brett A. Johnson; Xiaoxia Qi; John McAnally; Joseph A. Hill; James A. Richardson; Rhonda Bassel-Duby; Eric Olson; The Endothelial-Specific MicroRNA miR-126 Governs Vascular Integrity and Angiogenesis. Developmental Cell 2008, 15, 261-271, 10.1016/j.devcel.2008.07.002.
- Jason E. Fish; Massimo Mattia Santoro; Sarah U. Morton; Sangho Yu; Ru-Fang Yeh; Joshua Wythe; Kathryn N Ivey; Benoit G. Bruneau; Didier Yr Stainier; Deepak Srivastava; et al. miR-126 Regulates Angiogenic Signaling and Vascular Integrity. Developmental Cell 2008, 15, 272-284, 10.1016/j.devcel.2008.07.008.
- Roberto Sessa; Giorgio Seano; Laura Di Blasio; Paolo Armando Gagliardi; Claudio Isella; Enzo Medico; Franco Cotelli; F. Bussolino; Luca Primo; The miR-126 regulates Angiopoietin-1 signaling and vessel maturation by targeting p85β. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823, 1925-1935, 10.1016/j.bbamcr.2012.07.011.
- Coen Van Solingen; Leonard Seghers; Roel Bijkerk; Jacques M.G.J. Duijs; Marko K. Roeten; Annemarie M. Van Oeveren‐Rietdijk; Hans J. Baelde; Matthieu Monge; Joost Vos; Hetty C. De Boer; Paul H. A. Quax; Ton J. Rabelink; Anton Jan Van Zonneveld; Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. Journal of Cellular and Molecular Medicine 2008, 13, 1577-1585, 10.1111/j.1582-4934.2008.00613.x.
- Hai-Xiang Sun; D.-Y. Zeng; Ruo-Tian Li; Rui-Ping Pang; Hui Yang; Y.-L. Hu; Qun Zhang; Yue Jiang; Lin-Yan Huang; Yong-Bo Tang; Gui-Jun Yan; Jia-Guo Zhou; Essential Role of MicroRNA-155 in Regulating Endothelium-Dependent Vasorelaxation by Targeting Endothelial Nitric Oxide Synthase. Hypertension 2012, 60, 1407-1414, 10.1161/hypertensionaha.112.197301.
- F Muramatsu; Hiroyasu Kidoya; Hisamichi Naito; S Sakimoto; N Takakura; microRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene 2012, 32, 414-421, 10.1038/onc.2012.68.
- John Hung; Vladislav Miscianinov; Judith C. Sluimer; David E. Newby; Andrew H. Baker; Targeting Non-coding RNA in Vascular Biology and Disease. Frontiers in Physiology 2018, 9, 1655, 10.3389/fphys.2018.01655.
- Amankeldi Salybekov; Ainur K. Salybekova; Roberto Pola; Takayuki Asahara; Sonic Hedgehog Signaling Pathway in Endothelial Progenitor Cell Biology for Vascular Medicine. International Journal of Molecular Sciences 2018, 19, 3040, 10.3390/ijms19103040.
- Pan, Z.; Tian, Y.; Niu, G.; Cao, C. Role of microRNAs in remodeling the tumor microenvironment (Review). Int. J. Oncol. 2019.
- Kai Zhu; Qi Pan; Xin Zhang; Ling-Qun Kong; Jia Fan; Zhi Dai; Lu Wang; Xin-Rong Yang; Jie Hu; Jin-Liang Wan; Yi-Ming Zhao; Zhong-Hua Tao; Zong-Tao Chai; Hai-Ying Zeng; Zhao-You Tang; H. C. Sun; Jian Zhou; Patric Jansson; Fei Yue; Jing Sun; Daohai Zhang; Dong-Hun Bae; Sumit Sahni; Ying Zheng; Qian Zhao; Zaklina Kovacevic; Des R. Richardson; MiR-146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis 2013, 34, 2071-2079, 10.1093/carcin/bgt160.
- Thomas Wurdinger; Bakhos A. Tannous; Okay Saydam; Johan Skog; Stephan Grau; Jürgen Soutschek; Ralph Weissleder; Xandra O. Breakefield; Anna M. Krichevsky; miR-296 Regulates Growth Factor Receptor Overexpression in Angiogenic Endothelial Cells. Cancer Cell 2008, 14, 382-393, 10.1016/j.ccr.2008.10.005.
- Dominique Thuringer; Jonathan Boucher; Gaetan Jego; Nicolas Pernet; Laurent Cronier; Arlette Hammann; E. Solary; Carmen Garrido; Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 2016, 7, 73925-73934, 10.18632/oncotarget.12136.
- Zhicheng Zeng; Yuling Li; Yangjian Pan; Xiaoliang Lan; Fuyao Song; Jingbo Sun; Kun Zhou; Xiaolong Liu; Xiaoli Ren; Feifei Wang; Jinlong Hu; Xiaohui Zhu; Wei Yang; Wenting Liao; Guoxin Li; Yanqing Ding; L. Liang; Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nature Communications 2018, 9, 5395, 10.1038/s41467-018-07810-w.
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral Sphingomyelinase 2 (nSMase2)-dependent Exosomal Transfer of Angiogenic MicroRNAs Regulate Cancer Cell Metastasis. J. Biol. Chem. 2013, 288, 10849–10859.
- Yue-Chao Fan; Peng-Jin Mei; Chen Chen; Fa-An Miao; Hui Zhang; Zhong-Lin Li; MiR-29c inhibits glioma cell proliferation, migration, invasion and angiogenesis. Journal of Neuro-Oncology 2013, 115, 179-188, 10.1007/s11060-013-1223-2.
- Jong-Kuen Lee; Sae-Ra Park; Bong-Kwang Jung; Yoon-Kyung Jeon; Yeong-Shin Lee; Min-Kyoung Kim; Yong-Goo Kim; Ji-Young Jang; Chul-Woo Kim; Exosomes Derived from Mesenchymal Stem Cells Suppress Angiogenesis by Down-Regulating VEGF Expression in Breast Cancer Cells. PLOS ONE 2013, 8, e84256, 10.1371/journal.pone.0084256.
- Guanglei Zhuang; Xiumin Wu; Zhaoshi Jiang; Ian Kasman; Jenny Yao; Yinghui Guan; Jason Oeh; Zora Modrusan; Carlos Bais; Deepak Sampath; Napoleone Ferrara; Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. The EMBO Journal 2012, 31, 3513-3523, 10.1038/emboj.2012.183.
- Xu Chen; Fan Yang; Tianze Zhang; Wei Wang; Wenjin Xi; Yufang Li; Dan Zhang; Yi Huo; Jianning Zhang; Angang Yang; Tao Wang; MiR-9 promotes tumorigenesis and angiogenesis and is activated by MYC and OCT4 in human glioma. Journal of Experimental & Clinical Cancer Research 2019, 38, 99, 10.1186/s13046-019-1078-2.
- Haiou Yang; Haiyang Zhang; Shaohua Ge; Tao Ning; Ming Bai; Jialu Li; Shuang Li; Wu Sun; Ting Deng; Le Zhang; et al.Guoguang YingYi Ba Exosome-Derived miR-130a Activates Angiogenesis in Gastric Cancer by Targeting C-MYB in Vascular Endothelial Cells. Molecular Therapy 2018, 26, 2466-2475, 10.1016/j.ymthe.2018.07.023.
- Liang Liang; Lei Zhao; Ying Zan; Qing Zhu; Juan Ren; Xinhan Zhao; MiR-93-5p enhances growth and angiogenesis capacity of HUVECs by down-regulating EPLIN. Oncotarget 2017, 8, 107033-107043, 10.18632/oncotarget.22300.
- L Fang; Z Deng; T Shatseva; J Yang; C Peng; W W Du; A J Yee; L C Ang; C He; S W Shan; Burton B. Yang; MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-β8. Oncogene 2010, 30, 806-821, 10.1038/onc.2010.465.
- W Kong; L He; E J Richards; Sridevi Challa; C-X Xu; J Permuth-Wey; J M Lancaster; D Coppola; T A Sellers; J Y Djeu; et al.George Cheng Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2013, 33, 679-89, 10.1038/onc.2012.636.
- Guangmei Mao; Yan Liu; Xi Fang; Yahan Liu; Li Fang; Lianjun Lin; Xinmin Liu; Nanping Wang; Tumor-derived microRNA-494 promotes angiogenesis in non-small cell lung cancer. Angiogenesis 2015, 18, 373-382, 10.1007/s10456-015-9474-5.
- Xiangdong Liu; Xiang Gao; Wentao Zhang; Tianyi Zhu; Wei Bi; Yanrong Zhang; MicroRNA-204 deregulation in lung adenocarcinoma controls the biological behaviors of endothelial cells potentially by modulating Janus kinase 2-signal transducer and activator of transcription 3 pathway. IUBMB Life 2017, 70, 81-91, 10.1002/iub.1706.
- Zhang, L.; Lv, Z.; Xu, J.; Chen, C.; Ge, Q.; Li, P.; Wei, D.; Wu, Z.; Sun, X. Micro RNA -134 inhibits osteosarcoma angiogenesis and proliferation by targeting the VEGFA / VEGFR 1 pathway. FEBS J. 2018, 285, 1359–1371.
- Li-Hong Wang; Hsiao-Chi Tsai; Yu-Che Cheng; Chih-Yang Lin; Yuan-Li Huang; Chun-Hao Tsai; Guo-Hong Xu; Shih-Wei Wang; Yi-Chin Fong; Chih-Hsin Tang; CTGF promotes osteosarcoma angiogenesis by regulating miR-543/angiopoietin 2 signaling. Cancer Letters 2017, 391, 28-37, 10.1016/j.canlet.2017.01.013.
- Munekazu Yamakuchi; Craig D. Lotterman; Clare Bao; Ralph H. Hruban; Baktiar Karim; Joshua T. Mendell; David Huso; Charles J. Lowenstein; P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proceedings of the National Academy of Sciences 2010, 107, 6334-6339, 10.1073/pnas.0911082107.
- Jing-Jun Yan; Yu-Nan Zhang; Jia-Zhi Liao; Kun-Peng Ke; Ying Chang; Pei-Yuan Li; Min Wang; Ju-Sheng Lin; Xingxing He; MiR-497 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting VEGFA and AEG-1. Oncotarget 2015, 6, 29527-29542, 10.18632/oncotarget.5012.


