 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | François DUPUIS | + 1865 word(s) | 1865 | 2021-06-29 09:31:50 | | | |
| 2 | Conner Chen | Meta information modification | 1865 | 2021-07-05 10:11:20 | | |
Video Upload Options
Ang II is known as the main effector of the RAS. Ang II binds with a similar affinity to two receptors: the Ang II type 1 receptor (AT1) and type 2 receptor (AT2), both belonging to the G protein-coupled receptor (GPCR) family and sharing 50% homology and 34% identity in their amino acid sequence. Ang II activation of AT1 is acknowledged as triggering most of the known effects of RAS stimulation, such as vasoconstriction, water and sodium retention and aldosterone release by the adrenal glands. This leads to increases in blood pressure, cardiovascular remodeling and fibrosis. Due to its wide physiological effects, AT1 plays a critical role in many pathological conditions and cardiovascular diseases, like cardiac hypertrophy, hypertension and heart failure.
1. Implication of AT1 in Cerebrovascular Disease
1.1. Role of AT1 Activation in Strokes
Strokes are a major cause of death in industrialized countries and may lead to various physical disabilities consecutive to neurological deficit [1]. Strokes may result from two distinct etiologies: ischemia or hemorrhage. An ischemic stroke is the consequence of an obstruction or a vasospasm of a cerebral artery or arteriole [2], whereas a hemorrhagic stroke results from the rupture of the arterial, or arteriolar, wall. In both cases, the subsequent blood deprivation will lead to reduced oxygen levels, ischemia and necrosis of the downstream brain tissue.
Numerous preclinical data suggest that an overactivation of AT1 by Ang II might contribute to the severity of cerebral ischemia and its consequences, mainly through its vasoconstrictor effects on cerebral arteries [3]. Indeed, in a classic rodent model of ischemic stroke, the middle cerebral artery occlusion model (MCAo, in which the middle cerebral artery is temporarily occluded by a nylon monofilament), increases in the local production of Ang II and in the AT1-dependent vasoconstriction of cerebral arteries have been reported [4][5]. In the context of cerebral ischemia, Ang II and AT1 stimulation thus lead to a decreased perfusion of the penumbra and might expand the deleterious consequences of a stroke [6]. Besides these acute post-stroke effects, chronic AT1 stimulation is the main determinant of hypertension-induced inward remodeling of the cerebral arteriolar wall [7][8][9]. Indeed, hypertension, one of the major risk factors for strokes, induces a structural narrowing of the cerebral arterial lumen (thus reducing blood perfusion in case of a stroke) that only antihypertensive treatments targeting RAS and AT1 stimulation (ACE inhibitors and/or ARBs) are able to prevent [7][9][10].
Ang II and AT1 stimulation also induce a proinflammatory response [11] and increase oxidative stress through the production of ROS, thus promoting cellular damage and apoptosis [12][13]. These effects might also contribute to the increase in brain damage and the enlargement of the penumbra in case of ischemia and during reperfusion.
1.2. Role of AT1 Activation in Cerebral Aneurysm
Cerebral aneurysms are an abnormal condition in which a weakness of the cerebral arterial wall causes a localized dilation or bulge. Cerebral aneurysms can thus lead to hemorrhagic strokes showing a high mortality rate [14]. Given their role as major determinants of the cerebral arterial and arteriolar wall structure, RAS and AT1 might be good candidates to participate in the development of cerebral aneurysms.
Regarding abdominal aortic aneurysms, the involvement of Ang II and RAS stimulation has been clearly demonstrated in animals, and strong evidence have been gathered in humans [15][16]. However, when considering cerebral aneurysms, the role of RAS components and AT1 seems less obvious. In a study comparing patients with unruptured or ruptured cerebral aneurysm to controls, a significant drop in the mRNA expressions of ACE and AT1 has been observed [17]. Immunohistochemistry also revealed a decrease in AT1 and Ang II expression in unruptured aneurysmal walls. This decrease was even more pronounced in the arterial walls of patients with ruptured cerebral aneurysm [17]. Given the hypertrophic and remodeling effect of AT1, the authors suggested that a decreased expression of RAS components could lead to a reduced wall thickness and vascular remodeling which may result in cerebral aneurysms [17].
Preclinical data do not completely support these observations. In an experimental model of aneurysm in rats combining a left internal carotid artery ligation with a left renal artery ligation and a high salt diet (to increase blood pressure), no change in AT1 and ACE protein expression could be observed in cerebral aneurysmal walls. The authors therefore excluded any implication of AT1 or ACE in cerebral aneurysm formation [18][19], as further supported by the lack of effect of an ARB treatment [18]. If not involved in the development of cerebral aneurysms, RAS has been proposed to play a pivotal role in their rupture. In a mouse model of cerebral aneurysm (obtained by combining induced systemic hypertension with a single injection of elastase into the cerebrospinal fluid at the right basal cistern), Tada et al. prevented the spontaneous aneurysmal rupture with either an ACE inhibitor or an ARB, in a pressure-independent manner [20]. It is noteworthy that, unlike the rat model evoked above, this particular model of intracranial aneurysm showed an increased expression of Ang II and AT1 in aneurysmal walls, as revealed by immunohistochemistry [20].
The role of AT1 in the pathogenesis of cerebral aneurysm still appears to be controversial. Even if some data suggest a less consistent role of AT1 compared to other cerebrovascular diseases, further investigations are required to clarify the role of AT1 and its alternative signaling pathways in cerebral aneurysm.
1.3. Role of AT1 Activation in Traumatic Brain Injury
The Ang II/AT1 axis also plays a pivotal role in traumatic brain injury (TBI), another major cause of death and disability. A direct lesion of the brain will result in neuronal damage, increased cell death, dysfunction of the blood-brain barrier and increased inflammatory response. TBI may thus lead to motor and cognitive disorders. In victims of severe TBI, the presence of the D allele of ACE genotype, leading to increased ACE activity, has been correlated with disease mortality [21]. The subsequent excessive AT1 activation might contribute to neuronal injury and vulnerability by inducing cerebrovascular remodeling and inflammation [22].
Two genes encode for AT1 in rodents, AT1A and AT1B, and both share 93–94% amino-acids identity with the unique human AT1 protein [23][24]. AT1A has been described as the most abundant AT1 isoform expressed in mouse brain [25]. A decrease in susceptibility to traumatic injury was observed in AT1A knockout mice [26].
1.4. Role of AT1 Activation in Neurodegenerative Diseases
AT1 has also been reported as key determinant in many neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. However, the involvement of AT1 in these pathologies appears to be mostly located at the neuronal level. Recent reviews have been published on this matter [27][28][29] and this will not be further detailed here, as our present review addresses the cerebrovascular renin–angiotensin system.
1.5. Limitations
It is noteworthy that most of the studies cited above evaluated AT1 protein expression by means of antibodies targeting AT1 (Western blot and/or immunohistochemistry). These antibodies are notoriously unspecific [30][31][32] and the results obtained with these antibodies have therefore to be interpreted with caution. Nevertheless, the involvement of AT1 in cerebrovascular diseases has been proven by many other means.
The roles of AT1 in cerebrovascular diseases are summarized in Figure 1.
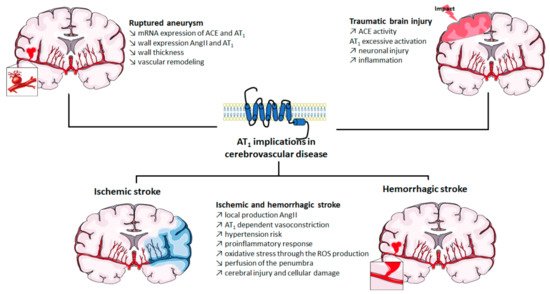
Figure 1. Implication of AT1 in cerebrovascular diseases. ACE: angiotensin-converting enzyme; Ang II: angiotensin II; AT1: angiotensin II type 1 receptor; mRNA: messenger ribonucleic acid; ROS: reactive oxygen species. The figure was prepared using the Servier Medical Art website (Creative Commons Attribution 3.0 unported License).
2. Blocking AT1 by ARBs in Cerebrovascular Disease: Success and Limitations
AT1 thus appears to be a first-order therapeutic target against cerebrovascular diseases, as has previously been clearly established for cardiovascular diseases. Currently, eight ARBs are clinically available, among which the most studied are losartan, candesartan, telmisartan and valsartan (Figure 2). Often described as surmountable (losartan) or unsurmountable (candesartan, telmisartan, valsartan) antagonists on the basis of their impact on the Ang II concentration response curve [33], ARBs show an inverse agonistic action by stabilizing the inactive state of AT1. ACE inhibitors and ARBs have been extensively studied to assess their potential preventive and curative effects in cerebrovascular pathologies.
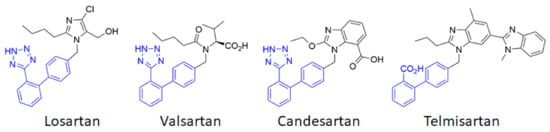
Figure 2. Chemical structures of the main angiotensin receptor blockers (ARB) used in therapeutics. The biphenyltetrazole (or the bioisosteric biphenyl carboxylic acid for telmisartan) is highlighted in blue.
Numerous preclinical studies show the beneficial effects of ARBs at the cerebrovascular level. ARBs regulate cerebral blood flow by blocking AT1 in cerebral arterial VSMC and endothelial cells [34]. ARBs improve blood perfusion, reduce apoptosis and decrease the infarct size after an ischemic stroke [35]. A 2-week pretreatment with candesartan before MCAo ischemic strokes in normotensive or hypertensive rats reduces the infarct volume and improves the lower and upper limits of cerebral blood flow autoregulation [36]. This chronic treatment also reduces vascular remodeling [37] and neuronal damage after cerebral ischemia [38][39]. Candesartan has also been reported to reduce neuronal damage in mice after a TBI [40] while valsartan could prevent these lesions [41]. Losartan has shown beneficial effects to prevent aneurysmal subarachnoid hemorrhage in a murine model [20]. Taken together, these preclinical data (which have recently been extensively reviewed [42][43]) provide evidence for the cerebrovascular potential of ARBs.
However, the transposition to clinic does not entirely support these preclinical data. Numerous clinical studies highlight the beneficial effect of long-term RAS blockades on stroke prevention. Early trials were conducted with ACE inhibitors. The stroke rate was reduced by 25% in patients receiving the ACE inhibitor captopril in the CAPPP (CAPtopril Prevention Project) trial [44] and this reduction reached 32% in the HOPE (Heart Outcome Prevention Evaluation) trial with the ACE inhibitor ramipril [45]. Similar drops in stroke recurrence were recorded in patients treated with perindopril, another ACE inhibitor, in the PROGRESS (perindopril PROtection aGainst REcurrent Stroke Study) study [46]. Interestingly, clinical studies using ARBs show identical results, suggesting that the beneficial effect of ACE inhibitors is mainly related to the prevention of AT1 stimulation. The LIFE (Losartan Intervention For Endpoint reduction in hypertension) study demonstrated that treatment with losartan prevented the risk of ischemia by 25% as compared to β-blockers that target β-adrenergic receptors [47]. The impact of candesartan was examined in the SCOPE (Study on COgnition and Prognosis in the Elderly) trial in which a risk reduction of 28% for non-fatal stroke was observed [48]. Similar results were found in the MOSES (MOrbidity and mortality after Stroke, Eprosartan compared with the calcium channel blocker nitrendipine for Secondary prevention) study with a 24% reduction in the stroke incidence in people treated with eprosartan [49]. Even if some studies failed to show such a dramatic effect (e.g., TRANSCEND [50] and PROFESS [51] trials), the prevention of strokes afforded by chronic ARBs treatment is well established. However, their efficacy in the acute and post-acute phases of strokes remains a matter of debate. The ACCESS (Acute Candesartan Cilexetil therapy in Stroke Survivors) study, in which candesartan therapy was initiated within 36 h after a stroke, revealed a lower mortality and a lower rate of vascular events after a one-year follow-up [52]. But a more recent trial failed to confirm these results. In the SCAST (Scandinavian Candesartan Acute Stroke Trial) study, candesartan treatment administered within 30 h after a stroke failed to show any beneficial effect when compared to placebo after 6 months [50][53]. In addition, ARBs act neither on cerebral lesions nor on the reduction in infarct volume, while this was observed in preclinical models. Thus, when considering acute stroke treatment, more in-depth studies should be carried out and the search for other therapeutic avenues that target AT1 should be considered.
References
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet 2014, 383, 245–255.
- Peisker, T.; Koznar, B.; Stetkarova, I.; Widimsky, P. Acute stroke therapy: A review. Trends Cardiovasc. Med. 2017, 27, 59–66.
- Näveri, L.; Strömberg, C.; Saavedra, J.M. Angiotensin II AT, receptor mediated contraction of the perfused rat cerebral artery. NeuroReport 1994, 5, 2278–2280.
- Stenman, E.; Edvinsson, L. Cerebral Ischemia Enhances Vascular Angiotensin AT 1 Receptor–Mediated Contraction in Rats. Stroke 2004, 35, 970–974.
- Kagiyama, T.; Kagiyama, S.; Phillips, M.I. Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regul. Pept. 2003, 110, 241–247.
- Walther, T.; Olah, L.; Harms, C.; Maul, B.; Bader, M.; Hörtnagl, H.; Schultheiss, H.-P.; Mies, G. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 2001, 16, 169–176.
- Dupuis, F.; Atkinson, J.; Limiñana, P.; Chillon, J.-M. Comparative effects of the angiotensin II receptor blocker, telmisartan, and the angiotensin-converting enzyme inhibitor, ramipril, on cerebrovascular structure in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 1061–1066.
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Cerebral Arteriolar Structure in Mice Overexpressing Human Renin and Angiotensinogen. Hypertension 2003, 41, 50–55.
- Dupuis, F.; Vincent, J.-M.; Limiñana, P.; Chillon, J.-M.; Capdeville-Atkinson, C.; Atkinson, J. Effects of suboptimal doses of the AT1 receptor blocker, telmisartan, with the angiotensin-converting enzyme inhibitor, ramipril, on cerebral arterioles in spontaneously hypertensive rat. J. Hypertens. 2010, 28, 1566–1573.
- Chillon, J.-M.; Baumbach, G. Effects of an Angiotensin-Converting Enzyme Inhibitor and a β-Blocker on Cerebral Arterioles in Rats. Hypertension 1999, 33, 856–861.
- Zhang, M.; Mao, Y.; Ramirez, S.; Tuma, R.; Chabrashvili, T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience 2010, 171, 852–858.
- Kazama, K.; Anrather, J.; Zhou, P.; Girouard, H.; Frys, K.; Milner, T.A.; Iadecola, C. Angiotensin II Impairs Neurovascular Coupling in Neocortex Through NADPH Oxidase–Derived Radicals. Circ. Res. 2004, 95, 1019–1026.
- De Silva, T.M.; Faraci, F.M. Effects of angiotensin II on the cerebral circulation: Role of oxidative stress. Front. Physiol. 2013, 3, 484.
- Inci, S.; Spetzler, R.F. Intracranial aneurysms and arterial hypertension: A review and hypothesis. Surg. Neurol. 2000, 53, 530–542.
- Daugherty, A.; Cassis, L. Angiotensin II and abdominal aortic aneurysms. Curr. Hypertens. Rep. 2004, 6, 442–446.
- Daugherty, A.; Rateri, D.L.; Cassis, L.A. Role of the Renin-Angiotensin System in the Development of Abdominal Aortic Aneurysms in Animals and Humans. Ann. N. Y. Acad. Sci. 2006, 1085, 82–91.
- Ohkuma, H.; Suzuki, S.; Fujita, S.; Nakamura, W. Role of a Decreased Expression of the Local Renin-Angiotensin System in the Etiology of Cerebral Aneurysms. Circulation 2003, 108, 785–787.
- Nishimura, M.; Aoki, T.; Kataoka, H.; Ishibashi, R.; Miyake, T.; Takagi, Y.; Morishita, R.; Hashimoto, N. Role of angiotensin II type 1 receptor in cerebral aneurysm formation in rats. Int. J. Mol. Med. 2009, 24, 353–359.
- Ishibashi, R.; Aoki, T.; Nishimura, M.; Miyamoto, S. Imidapril Inhibits Cerebral Aneurysm Formation in an Angiotensin-Converting Enzyme–Independent and Matrix Metalloproteinase-9–Dependent Manner. Neurosurgery 2012, 70, 722–730.
- Tada, Y.; Wada, K.; Shimada, K.; Makino, H.; Liang, E.I.; Murakami, S.; Kudo, M.; Kitazato, K.T.; Nagahiro, S.; Hashimoto, T. Roles of Hypertension in the Rupture of Intracranial Aneurysms. Stroke 2014, 45, 579–586.
- Kehoe, A.D.; Eleftheriou, K.I.; Heron, M.; Coats, T.J.; Montgomery, H. Angiotensin-converting enzyme genotype may predict survival following major trauma. Emerg. Med. J. 2008, 25, 759–761.
- Ando, H.; Zhou, J.; Macova, M.; Imboden, H.; Saavedra, J.M. Angiotensin II AT 1 Receptor Blockade Reverses Pathological Hypertrophy and Inflammation in Brain Microvessels of Spontaneously Hypertensive Rats. Stroke 2004, 35, 1726–1731.
- Iwai, N.; Inagami, T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992, 298, 257–260.
- Yamasaki, E.; Thakore, P.; Krishnan, V.; Earley, S. Differential expression of angiotensin II type 1 receptor subtypes within the cerebral microvasculature. Am. J. Physiol. Circ. Physiol. 2020, 318, H461–H469.
- Johren, O.; Saavedra, J.M. Expression of AT1A and AT1B angiotensin II receptor messenger RNA in forebrain of 2-wk-old rats. Am. J. Physiol. Metab. 1996, 271, E104–E112.
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315.
- Kalra, J.; Prakash, A.; Kumar, P.; Majeed, A.B.A. Cerebroprotective effects of RAS inhibitors: Beyond their cardio-renal actions. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 459–468.
- Gironacci, M.; Vicario, A.; Cerezo, G.; Silva, M.G. The depressor axis of the renin–angiotensin system and brain disorders: A translational approach. Clin. Sci. 2018, 132, 1021–1038.
- Villapol, S.; Saavedra, J.M. Neuroprotective Effects of Angiotensin Receptor Blockers. Am. J. Hypertens. 2015, 28, 289–299.
- Benicky, J.; Hafko, R.; Sanchez-Lemus, E.; Aguilera, G.; Saavedra, J.M. Six Commercially Available Angiotensin II AT1 Receptor Antibodies are Non-specific. Cell. Mol. Neurobiol. 2012, 32, 1353–1365.
- Herrera, M.; Sparks, M.A.; Alfonso-Pecchio, A.R.; Harrison-Bernard, L.M.; Coffman, T.M. Lack of Specificity of Commercial Antibodies Leads to Misidentification of Angiotensin Type 1 Receptor Protein. Hypertension 2013, 61, 253–258.
- Bouressam, M.-L.; Lartaud, I.; Dupuis, F.; Lecat, S. No answer to the lack of specificity: Mouse monoclonal antibody targeting the angiotensin II type 1 receptor AT1 fails to recognize its target. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 883–889.
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50.
- Zhou, J.; Pavel, J.; Macova, M.; Yu, Z.-X.; Imboden, H.; Ge, L.; Nishioku, T.; Dou, J.; Delgiacco, E.; Saavedra, J.M. AT 1 Receptor Blockade Regulates the Local Angiotensin II System in Cerebral Microvessels From Spontaneously Hypertensive Rats. Stroke 2006, 37, 1271–1276.
- Lou, M.; Blume, A.; Zhao, Y.; Gohlke, P.; Deuschl, G.; Herdegen, T.; Culman, J. Sustained Blockade of Brain AT1 Receptors before and after Focal Cerebral Ischemia Alleviates Neurologic Deficits and Reduces Neuronal Injury, Apoptosis, and Inflammatory Responses in the Rat. Br. J. Pharmacol. 2004, 24, 536–547.
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT1Blockade Normalizes Cerebrovascular Autoregulation and Reduces Cerebral Ischemia in Spontaneously Hypertensive Rats. Stroke 2000, 31, 2478–2486.
- Ito, T.; Yamakawa, H.; Bregonzio, C.; Terrón, J.A.; Falcón-Neri, A.; Saavedra, J.M. Protection Against Ischemia and Improvement of Cerebral Blood Flow in Genetically Hypertensive Rats by Chronic Pretreatment With an Angiotensin II AT 1 Antagonist. Stroke 2002, 33, 2297–2303.
- Groth, W.; Blume, A.; Gohlke, P.; Unger, T.; Culman, J. Chronic pretreatment with candesartan improves recovery from focal cerebral ischaemia in rats. J. Hypertens. 2003, 21, 2175–2182.
- Stier, C.T.; Adler, L.A.; Levine, S.; Chander, P.N. Stroke prevention by losartan in stroke-prone spontaneously hypertensive rats. J. Hypertens. 1993, 11, 37–42.
- Villapol, S.; Yaszemski, A.K.; Logan, T.T.; Sanchez-Lemus, E.; Saavedra, J.M.; Symes, A.J. Candesartan, an Angiotensin II AT1-Receptor Blocker and PPAR-γ Agonist, Reduces Lesion Volume and Improves Motor and Memory Function After Traumatic Brain Injury in Mice. Neuropsychopharmacology 2012, 37, 2817–2829.
- Li, J.-M.; Mogi, M.; Iwanami, J.; Min, L.-J.; Tsukuda, K.; Sakata, A.; Fujita, T.; Iwai, M.; Horiuchi, M. Temporary Pretreatment With the Angiotensin II Type 1 Receptor Blocker, Valsartan, Prevents Ischemic Brain Damage Through an Increase in Capillary Density. Stroke 2008, 39, 2029–2036.
- Wanderer, S.; Grüter, B.E.; Strange, F.; Sivanrupan, S.; Di Santo, S.; Widmer, H.R.; Fandino, J.; Marbacher, S.; Andereggen, L. The Role of Sartans in the Treatment of Stroke and Subarachnoid Hemorrhage: A Narrative Review of Preclinical and Clinical Studies. Brain Sci. 2020, 10, 153.
- Arroja, M.M.C.; Reid, E.; McCabe, C. Therapeutic potential of the renin angiotensin system in ischaemic stroke. Exp. Transl. Stroke Med. 2016, 8, 1–14.
- Hansson, L.; Lindholm, L.H.; Niskanen, L.; Lanke, J.; Hedner, T.; Niklason, A.; Luomanmäki, K.; Dahlöf, B.; de Faire, U.; Mörlin, C.; et al. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP) randomised trial. Lancet 1999, 353, 611–616.
- Bosch, J.; Yusuf, S.; Pogue, J.; Sleight, P.; Lonn, E.; Rangoonwala, B.; Davies, R.; Ostergren, J.; Probstfield, J. Use of ramipril in preventing stroke: Double blind randomised trial. BMJ 2002, 324, 699.
- PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6105 individuals with previous stroke or transient ischaemic attack. Lancet 2001, 358, 1033–1041.
- Dahlöf, B.; Devereux, R.B.; Kjeldsen, S.E.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O.; et al. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): A randomised trial against atenolol. Lancet 2002, 359, 995–1003.
- Lithell, H.; Hansson, L.; Skoog, I.; Elmfeldt, D.; Hofman, A.; Olofsson, B.; Trenkwalder, P.; Zanchetti, A. The Study on Cognition and Prognosis in the Elderly (SCOPE). J. Hypertens. 2003, 21, 875–886.
- Schrader, J.; Lüders, S.; Kulschewski, A.; Hammersen, F.; Plate, K.; Berger, J.; Zidek, W.; Dominiak, P.; Diener, H.C. The MOSES Study Group Morbidity and Mortality After Stroke, Eprosartan Compared with Nitrendipine for Secondary Prevention. Stroke 2005, 36, 1218–1224.
- Yusuf, S.; Teo, K.; Anderson, C.S.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet 2008, 372, 1174–1183.
- Yusuf, S.; Diener, H.-C.; Sacco, R.L.; Cotton, D.; Ôunpuu, S.; Lawton, W.A.; Palesch, Y.; Martin, R.H.; Albers, G.W.; Bath, P.; et al. Telmisartan to Prevent Recurrent Stroke and Cardiovascular Events. N. Engl. J. Med. 2008, 359, 1225–1237.
- Schrader, J.; Lüders, S.; Kulschewski, A.; Berger, J.; Zidek, W.; Treib, J.; Einhäupl, K.; Diener, H.C.; Dominiak, P. The ACCESS Study. Stroke 2003, 34, 1699–1703.
- Sandset, E.C.; Bath, P.; Boysen, G.; Jatuzis, D.; Kõrv, J.; Lüders, S.; Murray, G.D.; Richter, P.S.; Roine, R.O.; Terént, A.; et al. The angiotensin-receptor blocker candesartan for treatment of acute stroke (SCAST): A randomised, placebo-controlled, double-blind trial. Lancet 2011, 377, 741–750.


