AT
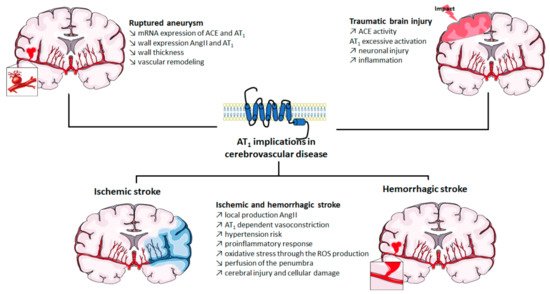
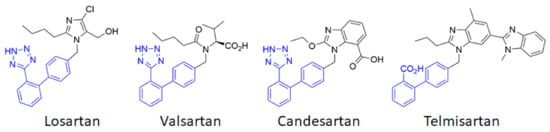
1 thus appears to be a first-order therapeutic target against cerebrovascular diseases, as has previously been clearly established for cardiovascular diseases. Currently, eight ARBs are clinically available, among which the most studied are losartan, candesartan, telmisartan and valsartan (
Figure 2). Often described as surmountable (losartan) or unsurmountable (candesartan, telmisartan, valsartan) antagonists on the basis of their impact on the Ang II concentration response curve [
51], ARBs show an inverse agonistic action by stabilizing the inactive state of AT
1. ACE inhibitors and ARBs have been extensively studied to assess their potential preventive and curative effects in cerebrovascular pathologies.
Numerous preclinical studies show the beneficial effects of ARBs at the cerebrovascular level. ARBs regulate cerebral blood flow by blocking AT
1 in cerebral arterial VSMC and endothelial cells [
52]. ARBs improve blood perfusion, reduce apoptosis and decrease the infarct size after an ischemic stroke [
53]. A 2-week pretreatment with candesartan before MCAo ischemic strokes in normotensive or hypertensive rats reduces the infarct volume and improves the lower and upper limits of cerebral blood flow autoregulation [
54]. This chronic treatment also reduces vascular remodeling [
55] and neuronal damage after cerebral ischemia [
56,
57]. Candesartan has also been reported to reduce neuronal damage in mice after a TBI [
58] while valsartan could prevent these lesions [
59]. Losartan has shown beneficial effects to prevent aneurysmal subarachnoid hemorrhage in a murine model [
40]. Taken together, these preclinical data (which have recently been extensively reviewed [
33,
60]) provide evidence for the cerebrovascular potential of ARBs.
However, the transposition to clinic does not entirely support these preclinical data. Numerous clinical studies highlight the beneficial effect of long-term RAS blockades on stroke prevention. Early trials were conducted with ACE inhibitors. The stroke rate was reduced by 25% in patients receiving the ACE inhibitor captopril in the CAPPP (CAPtopril Prevention Project) trial [
61] and this reduction reached 32% in the HOPE (Heart Outcome Prevention Evaluation) trial with the ACE inhibitor ramipril [
62]. Similar drops in stroke recurrence were recorded in patients treated with perindopril, another ACE inhibitor, in the PROGRESS (perindopril PROtection aGainst REcurrent Stroke Study) study [
63]. Interestingly, clinical studies using ARBs show identical results, suggesting that the beneficial effect of ACE inhibitors is mainly related to the prevention of AT
1 stimulation. The LIFE (Losartan Intervention For Endpoint reduction in hypertension) study demonstrated that treatment with losartan prevented the risk of ischemia by 25% as compared to β-blockers that target β-adrenergic receptors [
64]. The impact of candesartan was examined in the SCOPE (Study on COgnition and Prognosis in the Elderly) trial in which a risk reduction of 28% for non-fatal stroke was observed [
65]. Similar results were found in the MOSES (MOrbidity and mortality after Stroke, Eprosartan compared with the calcium channel blocker nitrendipine for Secondary prevention) study with a 24% reduction in the stroke incidence in people treated with eprosartan [
66]. Even if some studies failed to show such a dramatic effect (e.g., TRANSCEND [
67] and PROFESS [
68] trials), the prevention of strokes afforded by chronic ARBs treatment is well established. However, their efficacy in the acute and post-acute phases of strokes remains a matter of debate. The ACCESS (Acute Candesartan Cilexetil therapy in Stroke Survivors) study, in which candesartan therapy was initiated within 36 h after a stroke, revealed a lower mortality and a lower rate of vascular events after a one-year follow-up [
69]. But a more recent trial failed to confirm these results. In the SCAST (Scandinavian Candesartan Acute Stroke Trial) study, candesartan treatment administered within 30 h after a stroke failed to show any beneficial effect when compared to placebo after 6 months [
67,
70]. In addition, ARBs act neither on cerebral lesions nor on the reduction in infarct volume, while this was observed in preclinical models. Thus, when considering acute stroke treatment, more in-depth studies should be carried out and the search for other therapeutic avenues that target AT
1 should be considered.