Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alberto Ouro | + 14926 word(s) | 14926 | 2021-06-29 10:35:54 | | | |
| 2 | Rita Xu | -11866 word(s) | 3060 | 2021-06-30 07:49:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ouro, A. Ceramide Metabolism and Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/11465 (accessed on 07 February 2026).
Ouro A. Ceramide Metabolism and Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/11465. Accessed February 07, 2026.
Ouro, Alberto. "Ceramide Metabolism and Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/11465 (accessed February 07, 2026).
Ouro, A. (2021, June 29). Ceramide Metabolism and Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/11465
Ouro, Alberto. "Ceramide Metabolism and Parkinson’s Disease." Encyclopedia. Web. 29 June, 2021.
Copy Citation
Ceramide is a bioactive sphingolipid involved in numerous cellular processes. In addition to being the precursor of complex sphingolipids, ceramides can act as second messengers, especially when they are generated at the plasma membrane of cells. Its metabolic dysfunction may lead to or be a consequence of an underlying disease. Recent reports on transcriptomics and electrospray ionization mass spectrometry analysis have demonstrated the variation of specific levels of sphingolipids and enzymes involved in their metabolism in different neurodegenerative diseases. In the present review, we highlight the most relevant discoveries related to ceramide and neurodegeneration, with a special focus on Parkinson's disease.
ceramide
sphingolipids
Parkinson’s disease
neurodegeneration
sphingomyelinase
ceramide synthase
1. Introduction
Parkinson’s Disease (PD) is the second most common neurodegenerative disease [1]. PD affects 1% of the population over 60 years of age, with a higher risk of developing the disease in males [2][3]. The annual economic burden of PD in the European healthcare system per patient ranges from €2600 to €10,000 [4]. The disease presents with symptoms of motor impairment such as bradykinesia, rigidity, tremor, postural instability, and difficulty in speaking and swallowing. As for non-motor symptoms, patients present sleep disturbance, depression, cognitive impairment, sensory abnormalities, or autonomic dysfunction [1][2].
PD is characterized by the accumulation of misfolded α-synuclein (α-syn) in inclusions called Lewy bodies located in the substantia nigra of the central nervous system, resulting in the loss of dopaminergic neurons in substantia nigra pars compacta and striatal dopamine, which are responsible for the motor symptoms of the disease. Lewy bodies have also been found in other areas of the brain such as raphe nuclei, locus coeruleus, brainstem reticular formation, the dorsal motor nucleus of the vagus nerve, amygdala, hippocampus and nucleus basalis of Meynert, to which non-motor symptomatology is attributed [1][2].
As for the mechanisms responsible for PD, neuronal death and neurodegeneration have been linked to oxidative stress, vascular disfunction, tumor progression, altered mitochondrial, autophagy and proteolysis functions, inflammation, excitotoxicity and lysosomal storage disorders (LSD) [2][5]. In addition, it was reported that alterations in sphingolipid metabolism in the early stages of the disease may be linked to an increased risk of developing PD with dementia, while regulating the levels of certain sphingolipids by enzymatic regulation can slow the development of the disease [6].
Sphingolipids are widely distributed in the organism, including the central and peripheral nervous system [7][8]. The closest association of sphingolipids with neurodegenerative diseases was related to their structural function, especially the glycosphingolipids, as the main component of the plasma membrane of oligodendrocytes and myelin [9]. However, several studies have demonstrated the implication of sphingolipids in several key biological processes such as cellular proliferation and migration, differentiation, autophagy, apoptosis, senescence, and inflammation [8][10][11][12][13][14]. Recent works on transcriptomics and electrospray ionization mass spectrometry analysis (sphingolipidomics) have demonstrated the variation of specific levels of sphingolipids and enzymes involved in their metabolism in different neurodegenerative diseases [15][16][17].
Ceramides (Cers) have a dual role in cell biology since they are precursors of complex sphingolipids and second messengers to regulate cell homeostasis [7][8]. Ceramide (Cer) is highly expressed in neurons and modulates neuronal signaling, synaptic transmission, cell metabolism, neuron-glia interaction and cell survival [18][19][20]. The intracellular accumulation of Cer has been noticed as a critical step to neurodegeneration [21]. Neurodegenerative diseases and aging have a direct connection with oxidative stress. The increase in oxidative stress induces stimulation of the sphingomyelinase (SMase) activity and the consequent elevation of intracellular Cer concentration in neurons and oligodendrocytes [21][22]. Furthermore, mutations in different enzymes involved in the metabolism of Cer have been implicated in the development of neurodegenerative diseases. It should be noted that part of lipid metabolism takes place in lysosomal compartments. Thus, it is important to discriminate between lysosomal enzyme alterations, which would be part of LSD and non-lysosomal enzymes [23].
2. Sphingolipid Metabolism
Cers are considered the central hub of sphingolipid metabolism and can regulate key metabolic functions. In particular, Cers are potent inducers of cell cycle arrest and apoptotic cell death [13][24], and are implicated in inflammatory responses related to microbial infection, asthma, cardiovascular diseases or chronic obstructive pulmonary disease (COPD) [25][26]. Moreover, an unbalance of intracellular Cer levels can lead to neurological and neuroinflammatory diseases. Cers are synthesized mainly by three major pathways. Furthermore, Cer can be synthesized by the dephosphorylation of ceramide 1-phosphate (C1P) by ceramide 1-phosphate phosphatase (CPP). In addition to producing Cer, the metabolites of complex sphingolipid catabolism lead to the production of another bioactive molecules, such as sphingosine 1-phosphate (Figure 1).
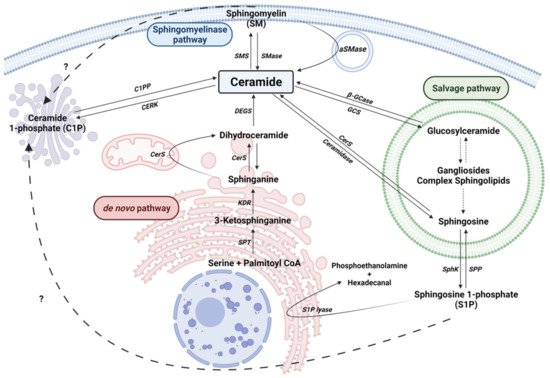
Figure 1. Sphingolipid metabolism. Solid arrows represent single reactions, whereas dashed arrows represent various step reactions. Interrogation marks with dashed arrows indicate unidentified mechanisms. Sphingomyelinase (SMase), sphingomyelin synthase (SMS), acid-sphingomyelinase (aSMase), Ceramide 1-phosphate phosphatase (C1PP), ceramide kinase (CERK), Serine palmitoyl transferase (SPT), 3-ketosphinganine reductase (KDR), Ceramide Synthase (CerS), sphingosine kinase (SphK), sphingosine 1-phosphate phosphatase (SPP), Sphingosine 1-phosphate lyase (S1P lyase) are represented by their acronyms.
2.1. The de novo Pathway
This pathway takes place in the endoplasmic reticulum (ER) where serine palmitoyltransferase (SPT) catalyzes the condensation of palmitate and serine to form 3-ketosphinganine (also called 3-ketodehydrosphingosine). Recent structural studies on SPT revealed a symmetrical dimer protein anchored to the ER membrane by six α-helices. The complex is formed by a single molecule of SPTLC1, SPTLC2, ssSPTa/b (two small subunits that enhance enzyme activity and also specify acyl-CoA substrate) and four regulatory subunits, ORMDLs (homologs of the yeast and plant Orms) [27]. Then, 3-keto-dihydrosphingosine reductase (KDR) produces sphinganine. Ceramide synthase (CerS) can then catalyze the formation of dihydroceramide (dhCer) through the incorporation of acyl-CoA of different chain lengths to sphinganine. There are six different isoforms of CerS (CerS1-6) identified in mammals and plants [28]. Specifically, CerS1 is highly expressed in the nervous system and skeletal muscles, but is almost undetectable in other types of tissue. CerS1 mainly generates 18 carbon chain Cer (C18-Cer), whereas CerS2 produces C22/24-Cer, CerS3 produces C26-Cer, CerS4 generates C18/20-Cer, CerS5 synthesizes C14/16-Cer and CerS6 forms C14/16-Cer. Cers with different acyl chain lengths have been detected in the mitochondria of the brain [29][30]. Furthermore, in brain tissue, CerS1, CerS2 and CerS6 enzymes were localized to the inner and outer mitochondrial membrane, and can induce the synthesis of C18-Cer, C22-Cer and C16-Cer, respectively [31]. Interestingly, mitochondrial CerS was associated with mitochondrial injury in cerebral ischemia/reperfusion with increased production of Cer [31]. The last step of this pathway is catalyzed by a dihydroceramide desaturase (DEGS), which introduces a double bond in position 4-5 trans of dhCer. DEGS is localized to the cytosolic leaflet of the ER membrane. In particular, genetic manipulation of the DEGS gene by tissue-specific deletion reduced hepatic steatosis and attenuated insulin resistance [32]. DEGS polymorphisms have been associated with the develop of cognitive impairment in schizophrenia [33]. Recently, DEGS mutation has been described to produce hypomyelination and degeneration of both central and peripheral nervous systems [34].
2.2. The Sphingomyelinase (SMase) Pathway
SMases are enzymes that hydrolyze sphingomyelin (SM) at the plasma membrane of cells to generate Cer and phosphocholine. SM hydrolysis is considered a fast mechanism for the production of Cer. There are five different types of SMases [35], and these have been classified according to their ion dependence, location and optimal pH. These include lysosomal and plasma membrane acid SMase (aSMase) [36], endoplasmic reticulum/nucleus and plasma membrane neutral Mg2+-dependent and neutral Mg2+-independent SMase (nSMase) [37], alkaline SMase (alkSMase), which is present in the intestinal tract and human bile [35][38], and a Zn2+-dependent secreted form of aSMase [39]. Meanwhile, aSMase and nSMase are implicated in cellular signaling, whereas alkSMase is implicated in the degradation of SM incorporated in the diet. It should be noted that aSMase and nSMase increase their activity by the action of pro-inflammatory stimuli, such as Tumor Necrosis Factor α (TNF-α), Interleukin-1β (IL-1β) or cytosolic phospholipase A2 (cPLA2), and it leads to elevation of intracellular Cer concentrations [40][41][42][43]. In addition, these enzymes are activated by some anticancer drugs, and by irradiation of cells with ultraviolet (UV) or ionizing radiation [44]. Mutations in the aSMase (SMPD1) gene results in disfunction of cholesterol and lipids metabolism, leading to Niemann-Pick’s disease [45][46].
There are different nSMase isoforms that have been characterized under different experimental settings [47]. The nSMase1 isoform (SPMD2 gene) is expressed in all cell types and highly enriched in the kidney. The nSMase2 isoform (SPMD3 gene) has a different domain structure than nSMase1. Contrary to nSMase 2, nSMase1 has two transmembrane domains, and instead has one collagen-like domain and two hydrophobic domains [48]. Of interest, nSMase2 is highly expressed in brain tissue [49]. Lastly, nSMase3 (SMPD4 gene) is ubiquitously expressed in all cell types. All of these SMase isoforms are Mg2+-dependent for expression of their activity. Dysregulation or stimulation of nSMase activity has been related to PD, Alzheimer’s disease, cognitive dysfunction or cerebral ischemia recovery [47][48][50][51][52].
2.3. The Salvage Pathway
This pathway involves a series of catabolic reactions that result in degradation of complex sphingolipids in acidic compartments, such as lysosomes. Complex sphingolipids, such as gangliosides (GM1, GM2 or GM3) or globosides, can be degraded to Lactosyl-ceramide (LacCer) by different reactions. Then, LacCer can be converted to Glucosyl-Ceramide (GlcCer) by LacCer hydrolase. Acid β-glucosidase 1 (β-GCase), encoded by GBA1 gene, converts GlcCer to lysosomal Cer. Deficiencies or dysfunction of this enzyme can lead to the accumulation of GlcCer and the development of the lysosomal storage disease known as Gaucher’s disease. Moreover, mutations in the GBA1 gene with loss of function have been linked to PD [53][54]. Contrarily, glucosylceramide synthase (GCS) transforms Cer into GlcCer. Once generated, Cer can be converted to sphingosine (Sph) by ceramidases. These enzymes differ in their optimal pH. There are three alkaline ceramidases (ACER1, ACER2 and ACER3), an acidic ceramidase (ASAH1) and a neutral ceramidase (ASAH2) [55][56]. ASAH1 is ubiquitously expressed in lysosomal compartments while ASAH2 is localized in plasma membranes, and mainly expressed in the small intestine and colon [55]. Sph is released to the cytosol and transformed to Cer by the activity of CerS in ER.
2.4. Ceramide Kinase/Ceramide 1-Phosphate Phosphatase (CerK/CPP) and Sphingosine Kinase/Sphingosine 1-Phosphate Phosphatase (SphK/SPP) Axis
Another relevant enzyme is Ceramide kinase (CerK), which phosphorylates Cer to produce Ceramide 1-phosphate (C1P). C1P is a key regulator of cell proliferation, survival and inflammation. The signaling pathways involved in C1P actions include mitogen-activated protein kinase kinase (MEK)/extracellularly regulated kinases (ERKs) 1/2, the mammalian target of rapamycin (mTOR), phosphatidylinositol 3-kinase (PI3K)/Akt, or protein kinase C-α [57][58][59], c-Jun N-terminal kinase (JNK) [60], or stimulation of vascular endothelial cell growth factor (VEGF) secretion [61]. Additionally, C1P-promoted cell survival implicates upregulation of inducible nitric oxide synthase (iNOS) expression [62], direct inhibition of aSMase [63][64] or SPT [65] and activation of the PI3K/Akt pathway [66]. It should be noted that C1P is implicated in the regulation of autophagy [67].
Sph that is released from the lysosomes to the cytosol in the Salvage pathway can be phosphorylated by sphingosine kinase (SK) to form S1P. It is one of the most studied sphingolipids. S1P has been described as a potent regulator of inflammatory processes through its union with specific membrane receptors. Thus far, five S1P receptors (S1PR1-5) have been described [68]. It is worth highlighting its involvement in glia activation processes, ischemic stroke and inflammatory processes in the vascular endothelium [69][70][71].
Dephosphorylation of S1P to Sph is due to the activity of S1P phosphatase (SPP) or by lipid phosphate phosphatase (LPP) activity [13]. However, S1P can be catalyzed by S1P lyase to produce hexadecenal and phosphoethanolamine in ER [72]. Of interest, it was observed, in both in vitro and in vivo models with a lack of S1P lyase an accumulation of β-amyloid and α-syn, promoting dysfunction of neuronal autophagy. In addition, the treatment with phosphoethanolamine restored autophagy, decreasing the deposits of β-amyloid and α-syn [73].
3. Neurodegeneration and Sphingolipid Metabolism
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease. It is characterized by extracellular deposits of β-amyloid (previously cleaved by secretases), called senile plaques, and intracellular build-up of hyperphosphorylated Tau protein in neurofibrillary tangles [74][75]. High levels of different species of Cer have been found in human samples from AD patients [76][77][78]. In addition, senile plaques were enriched in C18:1/18:0 and C18:1/20:0-Cer [79]. Likewise, aSMase and nSMase2 were found overexpressed in AD brain samples, correlated with increased Cer levels in blood [77]. Moreover, treatment with a cell-permeable analog of Cer (C6-Cer) or stimulation of endogenous Cer by nSMase activation stabilized β-site amyloid precursor protein cleaving enzyme 1 (BACE1) increasing β-amyloid accumulation [80]. Interestingly, β-amyloid has been reported to stimulate SMase activity in neurons [43][81][82], oligodendrocytes [83], dendritic [82] and endothelial cells [84], stimulating Cer accumulation and, thereby cell death. Additionally, overexpression of S1P lyase has been described to reduce β-amyloid production in N2a neuroblastoma cells [85]. Furthermore, it was observed a significant reduction of SphK1 and an increase of S1P lyase in AD human brain samples [86].
Different genetic diseases disrupt the metabolism of several molecules in the lysosomes, knowns as Lysosomal storage diseases (LSD). One of the main causes is lipid metabolism dysfunction, due to the alteration of enzymes such as aSMase or β-GCase [23][87][88][89]. LSD include different diseases, such as Niemann-Pick’s disease, Gaucher’s disease, Farber’s disease, Krabbe’s disease, Fabry’s disease, Tay-Sach’s disease, Sandhoff’s disease and ganglioside synthase deficiency. Lysosomal lipid storage occurs in all types of the disease, again highlighting the link between altered sphingolipid metabolism and neurodegeneration.
Niemann-Pick’s disease is a genetic disease that can be caused by two different types of mutations. Mutations in the SMPD1 gene lead to build-up of SM and the develop of Niemann-Pick’s disease type A and B [45][90]. Meanwhile, mutations in NPC Intracellular Cholesterol Transporter 1 or 2 (NPC1 or NPC2) alter cellular cholesterol trafficking and lipid metabolism disruption, leading to Niemann-Pick’s disease type C1 and C2 [91]. Recently, Torres et al. have shown that ASAH1 is downregulated in patients with Niemann-Pick’s disease type C1 [92]. They have also observed that the overexpression of ASAH1 improves mitochondrial function and reduces oxidative stress by decreasing STARD1.
Gaucher’s disease is due to a mutation in the gene encoding β-GCase (GBA), resulting in a deficit of the lysosomal enzyme, leading to an accumulation of GlcCer mainly in macrophages [53]. Elevated levels of glucosylsphingosine (GlcSph) were also found in the brain and were correlated with the phenotype of the disease [93]. Gaucher disease is associated with an increased risk of PD and dementia, since GBA deficiency increase α-syn aggregates [5].
Faber’s disease is caused by mutations in the ASAH1 gene, leading to an accumulation of Cer and cerebral atrophy. Interestingly, a rare epileptic disorder known as spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) is also associated with ASAH1 deficit [94]. Recently, C26-Cer was proposed as a biomarker for Faber’s disease diagnosis [95].
Krabbe’s disease is a genetic disease characterized by extensive demyelination, apoptosis of oligodendrocytes and Schwann cells and neurodegeneration due to mutations in the GALC gene that encode for galactocerebrosidase [96]. Recently, the possible link of greater cognitive impairment in PD patients with mutations in GALC gene was evidenced [97].
Huntington’s disease is a neurodegenerative disease strongly correlated with the expansion of CAG trinucleotide repeat within the huntingtin gene (HTT). It is characterized by progressive neurodegeneration and cognitive, motor and behavioral disturbances. Different studies carried out in in vivo models of Huntington’s disease have discovered a dysregulation in ganglioside metabolism [98][99]. Furthermore, a recent work has described a downregulation of SPT and CerS in mouse models, with a decrease in dihydroSphingosine, dihydroSphingosine-1-phosphate and dihydroCeramide (C18) [100].
Multiple sclerosis, also known as encephalomyelitis disseminate, is a chronic inflammatory disorder of the central nervous system characterized by demyelination and subsequent degeneration leading to neuronal damage and axonal loss. Its underlying etiology is unknown; however, genetic and environmental risks related to its development have been described [101]. CerS2 was found upregulated in monocytes and neutrophils isolated from mouse models [102]; meanwhile, CerS6 was increased in monocytes/macrophages [103][104]. Their overexpression has been associated with an increase in granulocyte stimulating factor (G-CSF)-induced C-X-C Motif Chemokine Receptor 2 (CXCR2) expression [103]. Additionally, downregulation of CerS2 and CerS6 were shown to inhibit the migration capacity of macrophages and neutrophils [103][104]. Therefore, CerS2 and CerS6 may represent a promising target for multiple sclerosis treatment. Moreover, plasma levels of C16-Cer, C24:1-Cer, C16-GlcCer and C24:1-GlcCer were increased and C16-LacCer was decreased in multiple sclerosis patients compared to healthy controls [105]. Furthermore, increased levels of C16:0- and C24:0-Cer were found in the cerebrospinal fluid samples from patients with multiple sclerosis [106].
Vascular dysfunction has been associated with the risk of neurodegeneration [107]. Notably, cerebral ischemia has been linked to pro-inflammatory processes in endothelial cells and loss of the integrity of the blood–brain barrier [108][109]. Sphingolipid metabolism has been described as a key factor in the progression and prognosis of brain ischemia. SMS1 was expressed in a time-dependent manner with a decrease in the first 24 h and recuperation at 72 h after transient middle cerebral artery occlusion (tMCAO) in rats [110]. Additionally, mice lacking aSMase exhibited a reduction in the infarct size in tMCAO, related to a decrease in Cer levels [111]. Moreover, a recent study demonstrated that aSMase protects against mild focal cerebral ischemia [112]. In preclinical studies, the levels of ceramides were increased 24 h after tMCAO in the ipsilateral hemisphere, especially in long-chain Cers, and decreased in SM [113]. Furthermore, recent studies in stroke patients showed elevated levels of long-chain Cers, while S1P and very long-chain Cers were decreased. Interestingly, high levels of long-chain Cers were associated with poor outcome at 48–72 h [114][115].
Glioblastoma is the most common and aggressive malignant brain tumor diagnosed in adults. The sphingolipids metabolism has emerged as a potential target for tumor cancer [116]. SPT inhibition by myriocin or specific siRNA inhibited the proliferation of human U87MG glioblastoma cells [117]. Des1 inhibitors such as γ-tocotrienol, phenoxodiol, or celecoxib have been described to induce autophagy in T98G and U87MG glioblastoma cell lines by dhCer accumulation [118]. Furthermore, N-[(1R,2S)-2-hydroxy-1-hydroxymethyl-2-(2-tridecyl-1-cyclopropenyl)ethyl]octanamide (GT11), another specific inhibitor of Des1, has been found to activate autophagy and apoptosis of the human U87MG glioma cell line [119]. Additionally, treatment with tetrahydrocannabinol (THC) produced an alteration of the lipid composition in the endoplasmic reticulum and reduction of Des1 expression, promoting autophagy and apoptosis in human U87MG glioma cells [119]. Interestingly, a correlation between SphK1 and poor survival has been observed in a clinical study with patients with glioblastoma [120]. Moreover, specific inhibition of SphK1 or SphK2 resulted in a cell-cycle arrest in U-1242 and U-87MG glioblastoma [120]. In addition, chemical or transcriptional down-regulation of SphK1 induces apoptosis and suppresses the growth of human glioblastoma cells and xenografts [121].
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905.
- Dextera, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144.
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178.
- Von Campenhausen, S.; Winter, Y.; e Silva, A.R.; Sampaio, C.; Ruzicka, E.; Barone, P.; Poewe, W.; Guekht, A.; Mateus, C.; Pfeiffer, K.-P.; et al. Costs of Illness and Care in Parkinson’s Disease: An Evaluation in Six Countries. Eur. Neuropsychopharmacol. 2011, 21, 180–191.
- Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 3304.
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 1–11.
- Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends Endocrinol. Metab. 2017, 28, 506–518.
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays Biochem. 2020, 64, 579–589.
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. J. Neurochem. 2021, 156, 403–414.
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793.
- Ouro, A.; Arana, L.; Gangoiti, P.; Gomez-Muñoz, A. Role of Ceramide 1-Phosphate in the Regulation of Cell Survival and Inflammation. Biochemistry 2012, 4.
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis. 2010, 9, 1–12.
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; et al. Control of Metabolism and Signaling of Simple Bioactive Sphingolipids: Implications in Disease. Prog. Lipid Res. 2010, 49, 316–334.
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150.
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal Fluid Sphingolipids, β-Amyloid, and Tau in Adults at Risk for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2486–2494.
- Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117.
- Van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as Prognostic Biomarkers of Neurodegeneration, Neuroinflammation, and Psychiatric Diseases and Their Emerging Role in Lipidomic Investigation Methods. Adv. Drug Deliv. Rev. 2020, 159, 232–244.
- Gulbins, A.; Grassm, H.; Hoehn, R.; Wilker, B.; Soddemann, M.; Kohnen, M.; Edwards, M.J.; Kornhuber, J.; Gulbins, E. Regulation of Neuronal Stem Cell Proliferation in the Hippocampus by Endothelial Ceramide. Cell. Physiol. Biochem. 2016, 39, 790–801.
- Schultz, A.; Larsson, C. Ceramide Influences Neurite Outgrowth and Neuroblastoma Cell Apoptosis Regulated by Novel Protein Kinase C Isoforms. J. Neurochem. 2004, 89, 1427–1435.
- Cruciani-Guglielmacci, C.; López, M.; Campana, M.; le Stunff, H. Brain Ceramide Metabolism in the Control of Energy Balance. Front. Physiol. 2017, 8, 787.
- Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and Neurodegeneration: Susceptibility of Neurons and Oligodendrocytes to Cell Damage and Death. J. Neurol. Sci. 2009, 278, 5–15.
- Pujol-Lereis, L.M. Alteration of Sphingolipids in Biofluids: Implications for Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3564.
- Platt, F.M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75.
- Watters, R.J.; Kester, M.; Tran, M.A.; Loughran, T.P.; Liu, X. Development and Use of Ceramide Nanoliposomes in Cancer. Methods Enzymol. 2012, 508, 89–108.
- Gomez-Muñoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordoñez, M. Control of Inflammatory Responses by Ceramide, Sphingosine 1-Phosphate and Ceramide 1-Phosphate. Prog. Lipid Res. 2016, 61, 51–62.
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191.
- Wattenberg, B.W. Kicking off Sphingolipid Biosynthesis: Structures of the Serine Palmitoyltransferase Complex. Nat. Struct. Mol. Biol. 2021, 28, 229–231.
- Kim, J.L.; Mestre, B.; Shin, S.-H.; Futerman, A.H. Ceramide Synthases: Reflections on the Impact of Dr. Lina M. Obeid. Cell. Signal. 2021, 82, 109958.
- Mignard, V.; Dubois, N.; Lanoé, D.; Joalland, M.P.; Oliver, L.; Pecqueur, C.; Heymann, D.; Paris, F.; Vallette, F.M.; Lalier, L. Sphingolipids Distribution at Mitochondria-Associated Membranes (MAM) upon Induction of Apoptosis. J. Lipid Res. 2020, 61, 1025–1037.
- Novgorodov, S.A.; Gudz, T.I. Ceramide and Mitochondria in Ischemic Brain Injury. Int. J. Biochem. Mol. Biol. 2011, 2, 347–361.
- Yu, J.; Novgorodov, S.A.; Chudakova, D.; Zhu, H.; Bielawska, A.; Bielawski, J.; Obeid, L.M.; Kindy, M.S.; Gudz, T.I. JNK3 Signaling Pathway Activates Ceramide Synthase Leading to Mitochondrial Dysfunction. J. Biol. Chem. 2007, 282, 25940–25949.
- Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392.
- Ohi, K.; Ursini, G.; Li, M.; Shin, J.H.; Ye, T.; Chen, Q.; Tao, R.; Kleinman, J.E.; Hyde, T.M.; Hashimoto, R.; et al. DEGS2 Polymorphism Associated with Cognition in Schizophrenia Is Associated with Gene Expression in Brain. Transl. Psychiatry 2015, 5, e550.
- Karsai, G.; Kraft, F.; Haag, N.; Korenke, G.C.; Hänisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-Associated Aberrant Sphingolipid Metabolism Impairs Nervous System Function in Humans. J. Clin. Investig. 2019, 129, 1229–1239.
- Goi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett. 2002, 531, 38–46.
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal Structure of Mammalian Acid Sphingomyelinase. Nat. Commun. 2016, 7, 1–9.
- Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, N.; Hannun, Y.A. The Extended Family of Neutral Sphingomyelinases. Biochemistry 2006, 45, 11247–11256.
- Cataldi, S.; Borrelli, A.; Ceccarini, M.R.; Nakashidze, I.; Codini, M.; Belov, O.; Ivanov, A.; Krasavin, E.; Ferri, I.; Conte, C.; et al. Acid and Neutral Sphingomyelinase Behavior in Radiation-Induced Liver Pyroptosis and in the Protective/Preventive Role of RMnSOD. Int. J. Mol. Sci. 2020, 21, 3281.
- Kornhuber, J.; Rhein, C.; Müller, C.P.; Mühle, C. Secretory Sphingomyelinase in Health and Disease. Biol. Chem. 2015, 396, 707–736.
- Jenkins, R.W.; Canals, D.; Idkowiak-Baldys, J.; Simbari, F.; Roddy, P.; Perry, D.M.; Kitatani, K.; Luberto, C.; Hannun, Y.A. Regulated Secretion of Acid Sphingomyelinase: Implications for Selectivity of Ceramide Formation. J. Biol. Chem. 2010, 285, 35706–35718.
- Becker, K.A.; Riethmuller, J.; Luth, A.; Doring, G.; Kleuser, B.; Gulbins, E. Acid Sphingomyelinase Inhibitors Normalize Pulmonary Ceramide and Inflammation in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 716–724.
- Gomez-Muñoz, A.; Gangoiti, P.; Arana, L.; Ouro, A.; Rivera, I.G.; Ordoñez, M.; Trueba, M. New Insights on the Role of Ceramide 1-Phosphate in Inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1060–1066.
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiol. Dis. 2006, 23, 178–189.
- Morad, S.A.F.; Cabot, M.C. Ceramide-Orchestrated Signalling in Cancer Cells. Nat. Rev. Cancer 2012, 13, 51–65.
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid Sphingomyelinase Deficient Mice: A Model of Types A and B Niemann–Pick Disease. Nat. Genet. 1995, 10, 288–293.
- Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 237–247.
- Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. NeuroMolecular Med. 2010, 12, 320–330.
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and Regulation of Neutral Sphingomyelinase-2 in Cellular and Pathological Processes. Adv. Biol. Regul. 2015, 57, 24–41.
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and Characterization of the Mammalian Brain-Specific, Mg2+-Dependent Neutral Sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900.
- Cataldi, S.; Arcuri, C.; Hunot, S.; Légeron, F.P.; Mecca, C.; Garcia-Gil, M.; Lazzarini, A.; Codini, M.; Beccari, T.; Tasegian, A.; et al. Neutral Sphingomyelinase Behaviour in Hippocampus Neuroinflammation of MPTP-Induced Mouse Model of Parkinson’s Disease and in Embryonic Hippocampal Cells. Mediat. Inflamm. 2017, 2017.
- Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.R.; Chu, M.; Haughey, N.J. Inhibition of Neutral Sphingomyelinase-2 Perturbs Brain Sphingolipid Balance and Spatial Memory in Mice. J. Neurosci. Res. 2010, 88, 2940–2951.
- Gu, L.Z.; Huang, B.S.; Shen, W.; Gao, L.; Ding, Z.Z.; Wu, H.W.; Guo, J. Early Activation of NSMase2/Ceramide Pathway in Astrocytes Is Involved in Ischemia-Associated Neuronal Damage via Inflammation in Rat Hippocampi. J. Neuroinflamm. 2013, 10, 1–16.
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Hum. Mutat. 2008, 29, 567–583.
- Velayati, A.; Yu, W.H.; Sidransky, E. The Role of Glucocerebrosidase Mutations in Parkinson Disease and Lewy Body Disorders. Curr. Neurol. Neurosci. Rep. 2010, 10, 190–198.
- Coant, N.; Hannun, Y.A. Neutral Ceramidase: Advances in Mechanisms, Cell Regulation, and Roles in Cancer. Adv. Biol. Regul. 2019, 71, 141–146.
- Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/Alkaline and Acid Ceramidase Activities Are Actively Released by Murine Endothelial Cells. Biochem. Biophys. Res. Commun. 2000, 275, 746–751.
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gomez-Muñoz, A.; Gomez-Munoz, A. Activation of Protein Kinase C-Alpha Is Essential for Stimulation of Cell Proliferation by Ceramide 1-Phosphate. FEBS Lett. 2010, 584, 517–524.
- Gangoiti, P.; Bernacchioni, C.; Donati, C.; Cencetti, F.; Ouro, A.; Gómez-Muñoz, A.; Bruni, P.; Gomez-Munoz, A.; Bruni, P. Ceramide 1-Phosphate Stimulates Proliferation of C2C12 Myoblasts. Biochimie 2012, 94, 597–607.
- Ouro, A.; Arana, L.; Gangoiti, P.; Rivera, I.G.; Ordoñez, M.; Trueba, M.; Lankalapalli, R.S.; Bittman, R.; Gomez-Muñoz, A. Ceramide 1-Phosphate Stimulates Glucose Uptake in Macrophages. Cell. Signal. 2013, 25, 786–795.
- Gangoiti, P.; Granado, M.H.; Wei, S.; Kong, J.Y.; Steinbrecher, U.P.; Gómez-muñoz, A. Ceramide 1-Phosphate Stimulates Macrophage Proliferation through Activation of the PI3-Kinase / PKB, JNK and ERK1 / 2 Pathways. Cell. Signal. 2008, 20, 726–736.
- Ouro, A.; Arana, L.; Riazy, M.; Zhang, P.; Gomez-Larrauri, A.; Steinbrecher, U.; Duronio, V.; Gomez-Muñoz, A. Vascular Endothelial Growth Factor Mediates Ceramide 1-Phosphate-Stimulated Macrophage Proliferation. Exp. Cell Res. 2017, 361, 277–283.
- Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gómez-Muñoz, A. Involvement of Nitric Oxide in the Promotion of Cell Survival by Ceramide 1-Phosphate. FEBS Lett. 2008, 582, 2263–2269.
- Gomez-Munoz, A.; Kong, J.; Salh, B.; Steinbrecher, U.P. Sphingosine-1-Phosphate Inhibits Acid Sphingomyelinase and Blocks Apoptosis in Macrophages. FEBS Lett. 2003, 539, 56–60.
- Newcomb, B.; Rhein, C.; Mileva, I.; Ahmad, R.; Clarke, C.J.; Snider, J.; Obeid, L.M.; Hannun, Y.A. Identification of an Acid Sphingomyelinase Ceramide Kinase Pathway in the Regulation of the Chemokine CCL5. J. Lipid Res. 2018, 59, 1219–1229.
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gómez-Muñoz, A. Ceramide 1-Phosphate Inhibits Serine Palmitoyltransferase and Blocks Apoptosis in Alveolar Macrophages. Biochim. Biophys. Acta 2009, 1791, 263–272.
- Gomez-Munoz, A.; Kong, J.Y.; Parhar, K.; Wang, S.W.; Gangoiti, P.; Gonzalez, M.; Eivemark, S.; Salh, B.; Duronio, V.; Steinbrecher, U.P. Ceramide-1-Phosphate Promotes Cell Survival through Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. FEBS Lett. 2005, 579, 3744–3750.
- Mishra, S.K.; Gao, Y.G.; Deng, Y.; Chalfant, C.E.; Hinchcliffe, E.H.; Brown, R.E. CPTP: A Sphingolipid Transfer Protein That Regulates Autophagy and Inflammasome Activation†. Autophagy 2018, 14, 862–879.
- Goetzl, E.J.; Wang, W.; McGiffert, C.; Huang, M.C.; Graler, M.H. Sphingosine 1-Phosphate and Its G Protein-Coupled Receptors Constitute a Multifunctional Immunoregulatory System. J. Cell Biochem. 2004, 92, 1104–1114.
- Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. NeuroMol. Med. 2021, 23, 211–223.
- Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-Phosphate Stimulates Proliferation and Migration of Satellite Cells: Role of S1P Receptors. Biochim. Biophys. Acta 2012, 1823, 439–450.
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial Sphingosine 1-Phosphate Receptors Promote Vascular Normalization and Antitumor Therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166.
- Saba, J.D. Fifty Years of Lyase and a Moment of Truth: Sphingosine Phosphate Lyase from Discovery to Disease. J. Lipid Res. 2019, 60, 456–463.
- Mitroi, D.N.; Karunakaran, I.; Gräler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (Sphingosine Phosphate Lyase 1) Modulates Neuronal Autophagy via Phosphatidylethanolamine Production. Autophagy 2017, 13, 885–899.
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36.
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293.
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B. Serum Ceramides Increase the Risk of Alzheimer Disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641.
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s Dis. 2012, 29, 537–547.
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455.
- Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and Sphingomyelinases in Senile Plaques. Neurobiol. Dis. 2014, 65, 193–201.
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide Stabilizes β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid β-Peptide Biogenesis. J. Biol. Chem. 2003, 278, 19777–19783.
- Desbène, C.; Malaplate-Armand, C.; Youssef, I.; Garcia, P.; Stenger, C.; Sauvée, M.; Fischer, N.; Rimet, D.; Koziel, V.; Escanyé, M.-C.; et al. Critical Role of CPLA2 in Aβ Oligomer-Induced Neurodegeneration and Memory Deficit. Neurobiol. Aging 2012, 33, 1123.e17–1123.e29.
- Jana, A.; Pahan, K. Fibrillar Amyloid-β-Activated Human Astroglia Kill Primary Human Neurons via Neutral Sphingomyelinase: Implications for Alzheimer’s Disease. J. Neurosci. 2010, 30, 12676–12689.
- Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β Peptide Induces Oligodendrocyte Death by Activating the Neutral Sphingomyelinase-Ceramide Pathway. J. Cell Biol. 2004, 164, 123–131.
- Yang, D.I.; Yeh, C.H.; Chen, S.; Xu, J.; Hsu, C.Y. Neutral Sphingomyelinase Activation in Endothelial and Glial Cell Death Induced by Amyloid Beta-Peptide. Neurobiol. Dis. 2004, 17, 99–107.
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci. 2011, 31, 6850–6857.
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced Sphingosine Kinase-1 and Enhanced Sphingosine 1-Phosphate Lyase Expression Demonstrate Deregulated Sphingosine 1-Phosphate Signaling in Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 1–10.
- Paciotti, S.; Albi, E.; Parnetti, L.; Beccari, T. Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease. J. Clin. Med. 2020, 9, 594.
- Zhao, Y.; Ren, J.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Lysosome Sorting of β-Glucocerebrosidase by LIMP-2 Is Targeted by the Mannose 6-Phosphate Receptor. Nat. Commun. 2014, 5, 1–12.
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. A Rare Lysosomal Enzyme Gene SMPD1 Variant (p.R591C) Associates with Parkinson’s Disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15.
- Conte, C.; Arcuri, C.; Cataldi, S.; Mecca, C.; Codini, M.; Ceccarini, M.R.; Patria, F.F.; Beccari, T.; Albi, E. Niemann-Pick Type a Disease: Behavior of Neutral Sphingomyelinase and Vitamin D Receptor. Int. J. Mol. Sci. 2019, 20, 2365.
- Vanier, M.T. Niemann-Pick diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 1717–1721.
- Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid Ceramidase Improves Mitochondrial Function and Oxidative Stress in Niemann-Pick Type C Disease by Repressing STARD1 Expression and Mitochondrial Cholesterol Accumulation. Redox Biol. 2021, 102052.
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine Accumulation in Tissues from Patients with Gaucher Disease: Correlation with Phenotype and Genotype. Mol. Genet. Metab. 2002, 76, 262–270.
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid Ceramidase Deficiency: Farber Disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 1–19.
- Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as Highly Sensitive Biomarker for the Diagnosis of Farber Disease. Sci. Rep. 2017, 7, 1–13.
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922.
- Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s Disease: The Potential Contribution of Galactosylceramidase Deficiency to Neuronal Vulnerability in Late-Onset Synucleinopathies. J. Neurosci. Res. 2016, 94, 1328–1332.
- Maglione, V.; Marchi, P.; di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired Ganglioside Metabolism in Huntington’s Disease and Neuroprotective Role of GM1. J. Neurosci. 2010, 30, 4072–4080.
- Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; et al. Disease-modifying Effects of Ganglioside GM1 in Huntington’s Disease Models. EMBO Mol. Med. 2017, 9, 1537–1557.
- Di Pardo, A.; Basit, A.; Armirotti, A.; Amico, E.; Castaldo, S.; Pepe, G.; Marracino, F.; Buttari, F.; Digilio, A.F.; Maglione, V. De Novo Synthesis of Sphingolipids Is Defective in Experimental Models of Huntington’s Disease. Front. Neurosci. 2017, 11, 698.
- Yamout, B.I.; Alroughani, R. Multiple Sclerosis. Semin. Neurol. 2018, 38, 212–225.
- Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreirós, N.; Henke, M.; et al. Lack of Ceramide Synthase 2 Suppresses the Development of Experimental Autoimmune Encephalomyelitis by Impairing the Migratory Capacity of Neutrophils. Brain Behav. Immun. 2015, 46, 280–292.
- Eberle, M.; Ebel, P.; Mayer, C.A.; Barthelmes, J.; Tafferner, N.; Ferreiros, N.; Ulshöfer, T.; Henke, M.; Foerch, C.; de Bazo, A.M.; et al. Exacerbation of Experimental Autoimmune Encephalomyelitis in Ceramide Synthase 6 Knockout Mice Is Associated with Enhanced Activation/Migration of Neutrophils. Immunol. Cell Biol. 2015, 93, 825–836.
- Schiffmann, S.; Ferreiros, N.; Birod, K.; Eberle, M.; Schreiber, Y.; Pfeilschifter, W.; Ziemann, U.; Pierre, S.; Scholich, K.; Grösch, S.; et al. Ceramide Synthase 6 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2012, 188, 5723–5733.
- Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshöfer, T.; Ferreirós, N.; Parnham, M.J.; Geisslinger, G.; et al. The Relevance of Ceramides and Their Synthesizing Enzymes for Multiple Sclerosis. Clin. Sci. 2018, 132, 1963–1976.
- Vidaurre, O.G.; Haines, J.D.; Sand, I.K.; Adula, K.P.; Huynh, J.L.; Mcgraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal Fluid Ceramides from Patients with Multiple Sclerosis Impair Neuronal Bioenergetics. Brain 2014, 137, 2271–2286.
- Sashindranath, M.; Nandurkar, H.H. Endothelial Dysfunction in the Brain: Setting the Stage for Stroke and Other Cerebrovascular Complications of Covid-19. Stroke 2021, 52, 1895–1904.
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Mohan, H.M.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral Ischemia Induces the Aggregation of Proteins Linked to Neurodegenerative Diseases. Sci. Rep. 2018, 8, 1–8.
- Kuźma, E.; Lourida, I.; Moore, S.F.; Levine, D.A.; Ukoumunne, O.C.; Llewellyn, D.J. Stroke and Dementia Risk: A Systematic Review and Meta-Analysis. Alzheimer’s Dement. 2018, 14, 1416–1426.
- Dmitrieva, V.G.; Torshina, E.V.; Yuzhakov, V.V.; Povarova, O.V.; Skvortsova, V.I.; Limborska, S.A.; Dergunova, L.V. Expression of Sphingomyelin Synthase 1 Gene in Rat Brain Focal Ischemia. Brain Res. 2008, 1188, 222–227.
- Yu, Z.F.; Nikolova-Karakashian, M.; Zhou, D.; Cheng, G.; Schuchman, E.H.; Mattson, M.P. Pivotal Role for Acidic Sphingomyelinase in Cerebral Ischemia-Induced Ceramide and Cytokine Production, and Neuronal Apoptosis. J. Mol. Neurosci. 2000, 15, 85–97.
- Hagemann, N.; Yusuf, A.M.; Martiny, C.; Zhang, X.; Kleinschnitz, C.; Gunzer, M.; Kolesnick, R.; Gulbins, E.; Hermann, D.M. Homozygous Smpd1 Deficiency Aggravates Brain Ischemia/ Reperfusion Injury by Mechanisms Involving Polymorphonuclear Neutrophils, Whereas Heterozygous Smpd1 Deficiency Protects against Mild Focal Cerebral Ischemia. Basic Res. Cardiol. 2020, 115, 1–14.
- Chao, H.C.; Lee, T.H.; Chiang, C.S.; Yang, S.Y.; Kuo, C.H.; Tang, S.C. Sphingolipidomics Investigation of the Temporal Dynamics after Ischemic Brain Injury. J. Proteome Res. 2019, 18, 3470–3478.
- Gui, Y.-K.; Li, Q.; Liu, L.; Zeng, P.; Ren, R.F.; Guo, Z.F.; Wang, G.H.; Song, J.G.; Zhang, P. Plasma Levels of Ceramides Relate to Ischemic Stroke Risk and Clinical Severity. Brain Res. Bull. 2020, 158, 122–127.
- Lee, T.H.; Cheng, C.N.; Chao, H.C.; Lee, C.H.; Kuo, C.H.; Tang, S.C.; Jeng, J.S. Plasma Ceramides Are Associated with Outcomes in Acute Ischemic Stroke Patients. J. Formos. Med. Assoc. 2021.
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111.
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochem. Pharmacol. 2015, 96, 119–130.
- Casasampere, M.; Ordóñez, Y.F.; Casas, J.; Fabrias, G. Dihydroceramide Desaturase Inhibitors Induce Autophagy via Dihydroceramide-Dependent and Independent Mechanisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 264–275.
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy of Cancer Cells via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229.
- Van Brooklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine Kinase-1 Expression Correlates with Poor Survival of Patients with Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705.
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting Sphingosine Kinase 1 Inhibits Akt Signaling, Induces Apoptosis, and Suppresses Growth of Human Glioblastoma Cells and Xenografts. Cancer Res. 2009, 69, 6915–6923.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
30 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No