Ceramide is a bioactive sphingolipid involved in numerous cellular processes. In addition to being the precursor of complex sphingolipids, ceramides can act as second messengers, especially when they are generated at the plasma membrane of cells. Its metabolic dysfunction may lead to or be a consequence of an underlying disease. Recent reports on transcriptomics and electrospray ionization mass spectrometry analysis have demonstrated the variation of specific levels of sphingolipids and enzymes involved in their metabolism in different neurodegenerative diseases. In the present review, we highlight the most relevant discoveries related to ceramide and neurodegeneration, with a special focus on Parkinson's disease.
- ceramide
- sphingolipids
- Parkinson’s disease
- neurodegeneration
- sphingomyelinase
- ceramide synthase
Review
Ceramide metabolism and Parkinson’s Disease - therapeutic targets
Antía Custodia1,+, Marta Aramburu-Núñez1,+, Clara Correa-Paz1, Adrián Posado-Fernández1, Ana Gómez-Larrauri2,3, José Castillo1, Antonio Gómez-Muñoz2, Tomás Sobrino1,* and Alberto Ouro1,*
|
Citation: Lastname, F.; Lastname, F.; Lastname, F. Title. Biomolecules 2021, 11, x. https://doi.org/10.3390/xxxxx Academic Editor: Firstname Lastname Received: date Accepted: date Published: date Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Copyright: © 2021 by the authors. Submitted for possible open access publication under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/). |

1 Clinical Neurosciences Research Laboratories, Health Research Institute of Santiago de Compostela (IDIS), Travesa da Choupana s/n, 15706, Santiago de Compostela, Spain
2 Department of Biochemistry and Molecular Biology. Faculty of Science and Technology. University of the Basque Country. P.O. Box 644, 48980, Bilbao. Spain
- Respiratory Department, Cruces University Hospital, Barakaldo, 48903, Bizkaia, Spain
+ These authors contributed equally to this work
* Correspondence author: alberto.ouro.villasante@sergas.es; Tel.: +34 664326589,
tomas.sobrino.moreiras@sergas.es; Tel. +34 981951098
Abstract: Ceramide is a bioactive sphingolipid involved in numerous cellular processes. In addition to being the precursor of complex sphingolipids, ceramides can act as second messengers, especially when they are generated at the plasma membrane of cells. Its metabolic dysfunction may lead to or be a consequence of an underlying disease. Recent reports on transcriptomics and electrospray ionization mass spectrometry analysis have demonstrated the variation of specific levels of sphingolipids and enzymes involved in their metabolism in different neurodegenerative diseases. In the present review, we highlight the most relevant discoveries related to ceramide and neurodegeneration, with a special focus on Parkinson's disease.
Keywords: ceramide, sphingolipids, Parkinson’s disease, neurodegeneration, sphingomyelinase, ceramide synthase, β-GCase, sphingolipidomics.
1. Introduction
Parkinson's Disease (PD) is the second most common neurodegenerative disease [1]. PD affects 1% of the population over 60 years of age, with a higher risk of developing the disease in males [2,3]. The annual economic burden of PD in the European healthcare system per patient ranges from €2,600 to €10,000 [4]. The disease presents with symptoms of motor impairment such as bradykinesia, rigidity, tremor, postural instability, difficulty in speaking and swallowing. As for non-motor symptoms, patients present sleep disturbance, depression, cognitive impairment, sensory abnormalities, or autonomic dysfunction [1,2].
The PD is characterized by the accumulation of misfolded α-synuclein (α-syn) in inclusions called Lewy bodies located in the substantia nigra of the central nervous system, resulting in the loss of dopaminergic neurons in substantia nigra pars compacta and striatal dopamine, which are responsible for the motor symptoms of the disease. Lewy bodies have also been found in other areas of the brain such as raphe nuclei, locus coeruleus, brainstem reticular formation, the dorsal motor nucleus of the vagus nerve, amygdala, hippocampus and nucleus basalis of Meynert, to which non-motor symptomatology is attributed [1,2].
As for the mechanisms responsible for PD, neuronal death and neurodegeneration have been linked to oxidative stress, vascular disfunction, tumour progression, altered mitochondrial, autophagy and proteolysis functions, inflammation, excitotoxicity and lysosomal storage disorders (LSD) [2,5]. In addition, it was reported that alterations in sphingolipid metabolism in the early stages of the disease may be linked to an increased risk of developing PD with dementia, while regulating the levels of certain sphingolipids by enzymatic regulation can slow the development of the disease [6].
Sphingolipids are widely distributed in the organism, including the central and peripheral nervous system [7,8]. The closest association of sphingolipids with neurodegenerative diseases was related to their structural function, especially the glycosphingolipids, as the main component of the plasma membrane of oligodendrocytes and myelin [9]. However, several studies have demonstrated the implication of sphingolipids in several key biological processes such as cellular proliferation and migration, differentiation, autophagy, apoptosis, senescence, and inflammation [8,10–14]. Recent works on transcriptomics and electrospray ionization mass spectrometry analysis (sphingolipidomics) have demonstrated the variation of specific levels of sphingolipids and enzymes involved in their metabolism in different neurodegenerative diseases [15–17].
Ceramides (Cers) have a dual role in cell biology since they are precursors of complex sphingolipids and second messengers to regulate cell homeostasis [8,18]. Ceramide (Cer) is highly expressed in neurons and modulates neuronal signaling, synaptic transmission, cell metabolism, neuron-glia interaction and cell survival [19–21]. The intracellular accumulation of Cer has been noticed as a critical step to neurodegeneration [22]. Neurodegenerative diseases and aging have a direct connection with oxidative stress. The increase in oxidative stress induces stimulation of the sphingomyelinase (SMase) activity and the consequent elevation of intracellular Cer concentration in neurons and oligodendrocytes [22,23]. Furthermore, mutations in different enzymes involved in the metabolism of Cer have been implicated in the development of neurodegenerative diseases. It should be noted that part of lipid metabolism takes place in lysosomal compartments. Thus, it is important to discriminate between lysosomal enzyme alterations, which would be part of LSD and non-lysosomal enzymes [24]. In this review, we will discuss the involvement of sphingolipids in neurodegeneration with special emphasis to PD.
2. Sphingolipid metabolism
Cers are considered the central hub of sphingolipid metabolism and can regulate key metabolic functions. In particular, Cers are potent inducers of cell cycle arrest and apoptotic cell death [13,25], and are implicated in inflammatory responses related to microbial infection, asthma, cardiovascular diseases or chronic obstructive pulmonary disease (COPD) [26,27]. Moreover, an unbalance of intracellular Cer levels can lead to neurological and neuroinflammatory diseases. Cers are synthesized mainly by three major pathways. Furthermore, Cer can be synthesized by the dephosphorylation of ceramide 1-phosphate (C1P) by ceramide 1-phosphate phosphatase (CPP). In addition to producing Cer, the metabolites of complex sphingolipid catabolism lead to the production of another bioactive molecules, such as sphingosine 1-phosphate (Fig. 1).
2.1 The de novo pathway
This pathway takes place in the endoplasmic reticulum (ER) where serine palmitoyltransferase (SPT) catalyzes the condensation of palmitate and serine to form 3-ketosphinganine (also called 3-ketodehydrosphingosine). Recent structural studies on SPT revealed a symmetrical dimer protein anchored to the ER membrane by six -helices. The complex is formed by a single molecule of SPTLC1, SPTLC2, ssSPTa/b (two small subunits that enhance enzyme activity and also specify acyl-CoA substrate) and four regulatory subunits, ORMDLs (homologs of the yeast and plant Orms) [28]. Then, 3-keto-dihydrosphingosine reductase (KDR) produces sphinganine. Ceramide synthase (CerS) can then catalyze the formation of dihydroceramide (dhCer) through the incorporation of acyl-CoA of different chain lengths to sphinganine. There are six different isoforms of CerS (CerS1-6) identified in mammals and plants [29]. Specifically, CerS1 is highly expressed in the nervous system and skeletal muscles but is almost undetectable in other types of tissue. CerS1 mainly generates 18 carbon chain Cer (C18-Cer), whereas CerS2 produces C22/24-Cer, CerS3 produces C26-Cer, CerS4 generates C18/20-Cer, CerS5 synthesizes C14/16-Cer and CerS6 forms C14/16-Cer. Cers with different acyl chain lengths have been detected in the mitochondria of the brain [30,31]. Furthermore, in brain tissue, CerS1, CerS2 and CerS6 enzymes were localized to the inner and outer mitochondrial membrane, and can induce the synthesis of C18-Cer, C22-Cer and C16-Cer, respectively [32]. Interestingly, mitochondrial CerS was associated with mitochondrial injury in cerebral ischemia/reperfusion with increased production of Cer [32]. The last step of this pathway is catalyzed by a dihydroceramide desaturase (DEGS), which introduce a double bond in position 4-5 trans of dhCer. DEGS is localized to the cytosolic leaflet of the ER membrane. In particular, genetic manipulation of DEGS gene by tissue specific deletion reduced hepatic steatosis and attenuated insulin resistance [33]. DEGS polymorphisms have been associated with the develop of cognitive impairment in schizophrenia [34]. Recently, DEGS mutation has been described to produce to hypomyelination and degeneration of both central and peripheral nervous systems [35].
2.2 The sphingomyelinase (SMase) pathway
SMases are enzymes that hydrolyze sphingomyelin (SM) at the plasma membrane of cells to generate Cer and phosphocholine. SM hydrolysis is considered a fast mechanism for the production of Cer. There are five different types of SMases [36], and these have been classified according to their ion dependence, location and optimal pH. These include lysosomal and plasma membrane acid SMase (aSMase) [37], endoplasmic reticulum/nucleus and plasma membrane neutral Mg2+-dependent and neutral Mg2+-independent SMase (nSMase) [38], alkaline SMase (alkSMase), which is present in the intestinal tract and human bile [39,40], and a Zn2+-dependent secreted form of aSMase [41]. Meanwhile, aSMase and nSMase are implicated in cellular signaling, whereas alkSMase is implicated in the degradation of SM incorporated in the diet. It should be noted that aSMase and nSMase increase their activity by the action of pro-inflammatory stimuli, such as Tumor Necrosis Factor α (TNF-α), Interleukin-1 (IL-1) or cytosolic phospholipase A2 (cPLA2), and it leads to elevation of intracellular Cer concentrations [42–45]. In addition, these enzymes are activated by some anticancer drugs, and by irradiation of cells with ultraviolet (UV) or ionizing radiation [46]. Mutations in aSMase (encoded by SMPD1) gene results in disfunction of cholesterol and lipids metabolism, leading to Niemann-Pick’s disease [47,48].
There are different nSMase isoforms that have been characterized under different experimental settings [49]. The nSMase1 isoform (SPMD2 gene) is expressed in all cell types and highly enriched in the kidney. The nSMase2 isoform (SPMD3 gene) has a different domain structure than nSMase1. Contrary to nSMase 2, nSMase1 has two transmembrane domains, and instead has one collagen-like domain and two hydrophobic domains [50]. Of interest, nSMase2 is highly expressed in brain tissue [51]. Lastly, nSMase3 (SMPD4 gene) is ubiquitously expressed in all cell types. All of these SMase isoforms are Mg2+-dependent for expression of their activity. Dysregulation or stimulation of nSMase activity has been related to PD, Alzheimer’s disease, cognitive dysfunction or cerebral ischemia recovery [49,50,52–54].
-
- The salvage pathway
This pathway involves a series of catabolic reactions that result in degradation of complex sphingolipids in acidic compartments, such as lysosomes. Complex sphingolipids, such as gangliosides (GM1, GM2 or GM3) or globosides, can be degraded to Lactosyl-ceramide (LacCer) by different reactions. Then LacCer can be converted to Glucosyl-Ceramide (GlcCer) by LacCer hydrolase. Acid β-glucosidase 1 (β-GCase), encoded by GBA1 gene, converts GlcCer to lysosomal Cer. Deficiencies or dysfunction of this enzyme can lead to the accumulation of GlcCer and the development of the lysosomal storage disease known as Gaucher's disease. Moreover, mutations in the GBA1 gene with loss of function have been linked to PD [55,56]. Contrary, glucosylceramide synthase (GCS) transforms Cer into GlcCer. Once generated, Cer can be converted to sphingosine (Sph) by ceramidases. These enzymes differ in their optimal pH. There are three alkaline ceramidases (ACER1, ACER2 and ACER3), an acidic ceramidase (ASAH1) and a neutral ceramidase (ASAH2) [57,58]. ASAH1 is ubiquitously expressed in lysosomal compartments while ASAH2 is localized in plasma membranes, and mainly expressed in the small intestine and colon [57]. Sph is released to the cytosol and transformed to Cer by the activity of CerS in ER.
-
- Ceramide Kinase / Ceramide 1-phosphate phosphatase (CerK/CPP) and Sphingosine Kinase / sphingosine phosphatase (SphK/SPP) axis
[59–61][62][63][64][65,66][67][68] It should be noted that C1P is implicated in the regulation of autophagy [69].
Sph that is released from the lysosomes to the cytosol in the Salvage pathway can be phosphorylated by sphingosine kinase (SK) to form S1P. It is one of the most studied sphingolipids. S1P has been described as a potent regulator of inflammatory processes through its union with specific membrane receptors. So far, five S1P receptors (S1PR1-5) have been described [70]. It is worth highlighting its involvement in glia activation processes, ischemic stroke and inflammatory processes in the vascular endothelium [71–73].
Dephosphorylation of S1P to Sph is due to the activity of S1P phosphatase (SPP) or by lipid phosphate phosphatase (LPP) activity [13]. However, S1P can be catalyzed by S1P lyase to produce hexadecenal and phosphoethanolamine in ER [74]. Of interest, it was observed, in both in vitro and in vivo models with a lack of S1P lyase an accumulation of β-amyloid and α-syn, promoting dysfunction of neuronal autophagy. In addition, the treatment with phosphoethanolamine restored autophagy, decreasing the deposits of β-amyloid and α-syn [75].
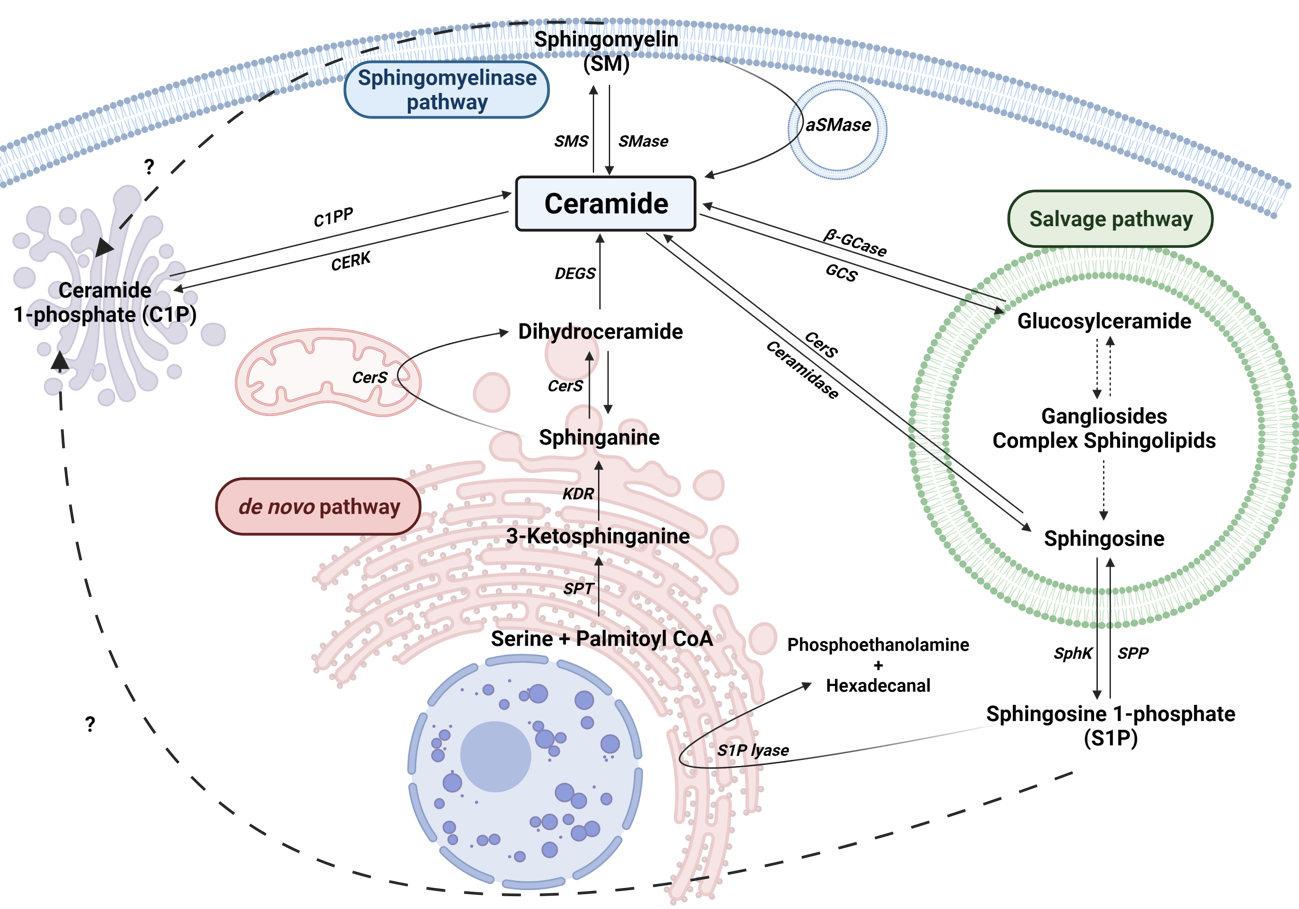
Figure 1. Sphingolipid metabolism. Solid arrows represent single reactions, whereas dashed arrows represent various step reactions. Interrogation marks with dashed arrows indicate unidentified mechanisms. Sphingomyelinase (SMase), sphingomyelin synthase (SMS), acid-sphingomyelinase (aSMase), Ceramide 1-phosphate phosphatase (C1PP), ceramide kinase (CERK), Serine palmitoyl transferase (SPT), 3-ketosphinganine reductase (KDR), Ceramide Synthase (CerS), sphingosine kinase (SphK), sphingosine 1-phosphate phosphatase (SPP), Sphingosine 1-phosphate lyase (S1P lyase) are represented by their acronyms.
2. Neurodegeneration and sphingolipid metabolism
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease. It is characterised by extracellular deposits of β-amyloid (previously cleaved by secretases), called senile plaques, and intracellular build-up of hyperphosphorylated Tau protein in neurofibrillary tangles [76,77]. High levels of different species of Cer have been found in human samples from AD patients [78–80]. In addition, senile plaques were enriched in C18:1/18:0 and C18:1/20:0-Cer [81]. Likewise, aSMase and nSMase2 were found overexpressed in AD brain samples, correlated with increased Cer levels in blood [82]. Moreover, treatment with a cell-permeable analog of Cer (C6-Cer) or stimulation of endogenous Cer by nSMase activation stabilized β-site amyloid precursor protein cleaving enzyme 1 (BACE1) increasing β-amyloid accumulation [83]. Interestingly, β-amyloid has been reported to stimulate SMase activity in neurons [84–86], oligodendrocytes [87], dendritic [86] and endothelial cells [88], stimulating Cer accumulation and, thereby cell death. Additionally, overexpression of S1P lyase has been described to reduce β-amyloid production in N2a neuroblastoma cells [89]. Furthermore, it was observed a significant reduction of SphK1 and an increase of S1P lyase in AD human brain samples [90].
Different genetic diseases disrupt the metabolism of several molecules in the lysosomes, knowns as Lysosomal storage diseases (LSD). One of the main causes is lipid metabolism dysfunction, due to the alteration of enzymes such as aSMase or β-GCase [91–94]. LSD include different diseases, such as Niemann-Pick’s disease, Gaucher’s disease, Farber’s disease, Krabbe’s disease, Fabry’s disease, Tay-Sach’s disease, Sandhoff’s disease and ganglioside synthase deficiency. Lysosomal lipid storage occurs in all types of the disease again highlighting the link between altered sphingolipid metabolism and neurodegeneration.
Niemann-Pick’s disease is a genetic disease that can be caused by two different types of mutations. Mutations in the SMPD1 gene lead to build-up of SM and the develop of Niemann-Pick’s disease type A and B [47,95]. Meanwhile, mutations in NPC Intracellular Cholesterol Transporter 1 or 2 (NPC1 or NPC2) alter cellular cholesterol trafficking and lipid metabolism disruption, leading to Niemann-Pick’s disease type C1 and C2 [96]. Recently, Torres et al. have shown that ASAH1 is downregulated in patients with Niemann-Pick’s disease type C1 [97]. They have also observed that the overexpression of ASAH1 improves mitochondrial function and reduces oxidative stress by decreasing STARD1.
Gaucher’s disease is due to a mutation in the gene encoding β-GCase (GBA), resulting in a deficit of the lysosomal enzyme, leading to an accumulation of GlcCer mainly in macrophages [55]. Elevated levels of glucosylsphingosine (GlcSph) were also found in the brain and were correlated with the phenotype of the disease [98]. Gaucher disease is associated with an increased risk of PD and dementia, since GBA deficiency increase α-syn aggregates [5].
Faber’s disease is caused by mutations in the ASAH1 gene, leading to an accumulation of Cer and cerebral atrophy. Interestingly, a rare epileptic disorder known as spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) is also associated with ASAH1 deficit [99]. Recently, C26-Cer was proposed as a biomarker for Faber’s disease diagnosis [100].
Krabbe’s disease is a genetic disease characterized by extensive demyelination, apoptosis of oligodendrocytes and Schwann cells and neurodegeneration due to mutations in the GALC gene that encode for galactocerebrosidase [101]. Recently, the possible link of greater cognitive impairment in PD patients with mutations in GALC gene was evidenced [102].
Huntington’s disease is a neurodegenerative disease strongly correlated with the expansion of CAG trinucleotide repeat within the huntingtin gene (HTT). It is characterized by progressive neurodegeneration and cognitive, motor and behavioural disturbances. Different studies carried out in in vivo models of Huntington’s disease have discovered a dysregulation in ganglioside metabolism [103,104]. Furthermore, a recent work has described a downregulation of SPT and CerS in mouse models, with a decrease in dihydroSphingosine, dihydroSphingosine-1-phosphate and dihydroCeramide (C18) [105].
Multiple sclerosis, also known as encephalomyelitis disseminate, is a chronic inflammatory disorder of the central nervous system characterised by demyelination and subsequent degeneration leading to neuronal damage and axonal loss. Its underlying aetiology is unknown, however genetic and environmental risks related to its development have been described [106]. CerS2 was found upregulated in monocytes and neutrophils isolated from mouse models [107], meanwhile, CerS6 was increased in monocytes/macrophages [108,109]. Their overexpression has been associated with an increase in granulocyte stimulating factor (G-CSF)-induced C-X-C Motif Chemokine Receptor 2 (CXCR2) expression [108]. Additionally, downregulation of CerS2 and CerS6 were shown to inhibit the migration capacity of macrophages and neutrophils [108,109]. Therefore, CerS2 and CerS6 may represent a promising target for multiple sclerosis treatment. Moreover, plasma levels of C16-Cer, C24:1-Cer, C16-GlcCer, and C24:1-GlcCer were increased and C16-LacCer was decreased in multiple sclerosis patients compared to healthy controls [110]. Furthermore, increased levels of C16:0- and C24:0-Cer were found in the cerebrospinal fluid samples from patients with multiple sclerosis [111].
Vascular dysfunction has been associated with the risk of neurodegeneration [112]. Notably, cerebral ischemia has been linked to pro-inflammatory processes in endothelial cells and loss of the integrity of the blood-brain barrier [113,114]. Sphingolipid metabolism has been described as a key factor in the progression and prognosis of brain ischemia. SMS1 was expressed in a time-dependent manner with a decrease in the first 24 hours and recuperation at 72 hours after transient middle cerebral artery occlusion (TMCAO) in rats [115]. Additionally, mice lacking aSMase exhibited a reduction in the infarct size in tMCAO, related to a decrease in Cer levels [116]. Moreover, a recent study demonstrated that aSMase protects against mild focal cerebral ischemia [117]. In preclinical studies, the levels of ceramides were increased 24 hours after tMCAO in the ipsilateral hemisphere, especially in long-chain Cers, and decreased in SM [118]. Furthermore, recent studies in stroke patients showed an elevated levels of long-chain Cers, while S1P and very long-chain Cers were decreased. Interestingly, high levels of long-chain Cers were associated with poor outcome at 48-72 hours [119,120].
Glioblastoma is the most common and aggressive malignant brain tumour diagnosed in adults. The sphingolipids metabolism has emerged as a potential target for tumour cancer [121]. SPT inhibition by myriocin or specific siRNA inhibited the proliferation of human U87MG glioblastoma cells [122]. Des1 inhibitors such as γ-tocotrienol, phenoxodiol, or celecoxib have been described to induce autophagy in T98G and U87MG glioblastoma cell lines by dhCer accumulation [123]. Furthermore, N-[(1R,2S)-2-hydroxy-1-hydroxymethyl-2-(2-tridecyl-1-cyclopropenyl)ethyl]octanamide (GT11) another specific inhibitor of Des1 have been found to activate autophagy and apoptosis of human U87MG glioma cell line [124]. Additionally, treatment with tetrahydrocannabinol (THC) produced an alteration of the lipid composition in the endoplasmic reticulum and reduction of Des1 expression promoting autophagy and apoptosis in human U87MG glioma cells [124]. Interestingly, a correlation between SphK1 and poor survival has been observed in a clinical study with patients with glioblastoma [125]. Moreover, specific inhibition of SphK1 or SphK2 resulted in a cell-cycle arrest in U-1242 and U-87MG glioblastoma [125]. In addition, chemical or transcriptional down-regulation of SphK1 induces apoptosis and suppresses the growth of human glioblastoma cells and xenografts [126].
3. Ceramide metabolism alterations in Parkinson’s Disease
The causes of PD are not completely understood and vary, from environmental factors such as exposure to toxins, as rotenone [127] or l‐methyl‐4‐phenyl‐ 1,2,3, 6‐tetrahydropyridine (MPTP) [128], to genetic factors. About 15 percent of patients with PD have a family history of the condition [3]. Family-linked cases can be a consequence of genetic modifications, considered as genetic risk factors, in some of the genes described so far, such as GBA, LRRK2, PLA2G6, PINK1 or SNCA gene.
3.1 GENETIC RISKS
3.1.1 GBA
GBA gene mutations are the most common autosomal dominant genetic cause of PD [129]. This gene encodes the lysosomal enzyme β-GCase, a lysosomal hydrolase that degrades GlcCer into Cer and glucose, and alternatively, degrades GlcSph and potentially other β-glucosides. Mutations in this gene that cause loss of expression or functionality in neurons or glia lead to Gaucher’s disease, belonging to Lysosomal Storage Diseases (LSD), by GlcCer accumulation [55,130,131]. Moreover, heterozygous mutations increase the lifetime risk to PD, since increased levels of GlcCer stimulate α-syn pathology [129,132,133]. In addition, recently, it was observed that in GBA mutant (N370S, L444P, KO) crossed with α-syn transgenic mouse there is a correlation between GlcSph accumulation and α-syn aggregation, indicating that GlcSph promotes α-syn assemblage [134]. Their interconnection is so tight that α-syn has also been shown to reduce β-GCase activity [135]. In fact, elevated levels of GlcCer can lead to α-syn accumulation and, conversely, α-syn can reduce β-GCase activity establishing a bidirectional pathogenic loop [135]. Recently, Jong Kim et al. showed that β-GCase deficiency leads to C18-Cer reduction and alters Rab8a location, a small cytosolic GTPase implicated in secretory autophagy [136]. In this work, the authors also demonstrated that ASAH1 inhibition increased C18-Cer and reduced GlcSph levels and oxidized α-syn [136]. The latter studies demonstrated the possible efficiency of a targeted therapy against GlcCer accumulation. Sardi et al. demonstrated that the use of a specific GCS inhibitor (GZ667161) in α-synucleinopathy transgenic mice reduced GlcCer and GlcSph levels, correlating with the reduction of α-syn aggregates and cognitive improvement [137].
A new small molecule (S-181) that increases β-GCase activity has recently been developed. In human induced pluripotent stem cell (iPSC)-derived dopaminergic neurons from PD patients carrying mutations in GBA, LRRK2, DJ-1 or PARKIN were found that S-181 reduced β-GCase activity. Furthermore, S-181 partially restored lysosomal function and consequently, reduced oxidized dopamine, GlcCer levels and α-syn accumulation. Moreover, S-181 treatment of Gba1D409V/- mice resulted in an increased wild-type β-GCase activity and consequent reduction of α-syn accumulation [138]. These results are in agreement with previous studies in which overexpression of β-GCase carried out by Adenovirus Vector (AAV) Gene Therapy observed a reduction of the α-syn aggregation [139,140].
Interestingly, GBA deficiency promotes a down-regulation of protein phosphatase 2A (PP2A) leading to an inhibition of autophagy and thus an accumulation of α-syn [141]. This result could be related to a stimulation of PP2A activity by Cer [142,143]. Additionally, it was observed that in lung epithelial cells, the pro-inflammatory cytokine TNF-α stimulates the concentration of intracellular Cer, promoting the stimulation of PP2A activity. Moreover, PP2A was described as modulator of c-Jun N-terminal kinases (JNK), extracellular signal-regulated kinase (ERK) and p38 pathways, stimulating the production of interleukin 8 (IL-8) and along with a pro-inflammatory cascade [144]. Therefore, a low concentration of Cer, due to a deficiency in GBA might produce a decrease in the activity of PP2A.
It is well established that α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) are the major receptors responsible for synaptic plasticity in neurons. In neurodegenerative diseases, this ability of neurons to adapt fails, leading to cognitive impairments [145]. Among all the proteins that regulate vesicular traffic and AMPAR activation in synapses, PP2A is a relevant enzyme. This phosphatase regulates the dephosphorylation of AMPARs and thus the inactivation and endocytosis of the receptors [146,147]. Furthermore, it has been reported that Cer reduces neuronal AMPAR levels in synapses after a treatment with pro-inflammatory molecules, such as IL-1β [148] or TNF-α [149]. These observations suggest that Cer regulates the activity of AMPARs through PP2A, although these aspects require further investigation.
Also of interest is the fact that Cer stimulates autophagy via inhibition of protein kinase B (PKB, also known as Akt) and activation of a mammalian tumour suppressor called Beclin-1 (BENC1), reducing the levels of Cer in GBA deficient neurons also produces a decrease in autophagy through this pathway [150].
3.1.2 LRRK2
LRRK2 is a member of the leucine-rich repeat kinase family of genes. Certain mutations associated with PD have been described, representing 1-2% of cases. Mutations in this autosomal dominant gene lead to an increase of LRRK2 activity [151]. Furthermore, different LRRK2 mutations have been associated with mitochondrial dysfunction, stimulation of reactive oxygen species (ROS) production, dysfunction in fission and fusion processes, mitophagy and dysregulation of cytoskeleton dynamics and mitochondria trafficking [152]. The predominant mutation is the modification of a glycine in the 2019 residue for a serine (LRRK2-G2019S) [153]. Recently, Boecker et al. demonstrated that hyperactivation of LRRK2 kinase activity increases phosphorylation of Rab GTPases and recruits the motor adaptor JNK-interacting protein 4 (JIP4) to the autophagosome membrane, leading to a disruption of autophagosome transport [154]. These observations were made in human iPSC-derived neurons gene-edited to express the G2019S mutation, and the results were reversed by genetic or pharmacological inhibition of LRRK2, making it a hopeful target for PD.
LRRK2 mutations have also been observed in patients with GBA mutations. However, the phenotype associated with carriers of individual GBA or LRRK2 mutations and the two joint mutations is not clear. A study with a cohort of 236 participants showed that the LRRK2 mutation together with GBA rescued from further mental degeneration [155]. Meanwhile, a recent study with a cohort of 1193 Ashkenazi Jewish population showed no differences between genotypes [156].
Ferrazza et al. demonstrated that LRRK2 activity is involved in the regulation of Cer metabolism. The authors observed that Lrrk2-/- transgenic mice showed increased β-GCase activity with elevated Cer levels in the brain [157]. Furthermore, increased β-GCase activity was determined in blood samples from patients with LRRK2-G2019S-PD [132]. This observation could be explained by the release of the lysosomal vesicles to the extracellular compartment [132]. Additionally, recent studies in neurons derived from PD patients with mutations in LRRK2 have shown that LRRK2 regulates β-GCase activity through the small GTPase Rab10. Moreover, treatment with specific LRRK2 inhibitors (in clinical phase trials) increased β-GCase activity improving cognition functions in PD patients [158].
It is well established that LRRK2 regulates mitochondrial homeostasis by Dynamin-related protein (Drp-1) and ROS production [159,160]. In addition, Drp-1 modifies Cer distribution in the outer membrane of the mitochondria, preventing mitophagy [161]. As mentioned above, there is a high expression of mitochondrial CerS in the brain [32]. Interestingly, genetic modifications of CerS in C. elegans have been linked to alterations in the inclusion of α-syn [162]. This result could be one of the explanations for the different species of Cer found in patient samples, in both blood and cerebrospinal fluid (CSF), as well as brain biopsies (elegantly reviewed by Pujol-Lereis [23]).
3.1.3 PLA2G6
PLA2G6 gene encodes a calcium-independent phospholipase A2 (iPLA2-VIA). This enzyme catalyzes the hydrolysis of glycerophospholipids, producing free fatty acids, including pro-inflammatory polyunsaturated arachidonic acid (AA), and lysophospholipids. Earlier, it was observed that mutations in this gene produce abnormal growth of neuronal axons [163], early-onset PD [164,165], and association with PD develop [164]. Additionally, it was also observed that these mutations caused dysregulation of Ca2+ homeostasis, leading to autophagic dysfunction and loss of dopaminergic neurons [166]. Interestingly, the loss of iPLA2-VIA results in a build-up of Cer [167], by a possible mechanism that alters the plasma composition. In fact, PLA2G6 binds to VPS35 (also mutated in some PD patients [168]) and VPS26, stimulating plasma membrane recycling. This process could stimulate the production of Cer in the lysosomes by aSMase. It should be noted that production of lysosomal Cer leads to α-syn accumulation [92].
Since Cer is the substrate for CerK, C1P concentration can be enhanced by increasing Cer levels [59–61]. [62][63][64][65,66][67][68]However, C1P levels in the blood of PD patients carrying the GBA mutations are reduced in comparison with non-carrier PD patients [169]. Nonetheless, the implication of CerK in PD requires further investigation
3.1.4 PINK1
The PINK1 gene encodes for phosphatase and tensin homolog (PTEN)-induced putative kinase protein 1 (PINK1), a mitochondrial-targeted serine/threonine kinase [170]. This protein is implicated in the maintenance of mitochondrial homeostasis. When mitochondrial membrane suffers a depolarization due to oxidative stress, PINK1 recruits the E3 ubiquitin ligase Parkin protein from the cytosol, initiating a process of mitochondrial degradation called mitophagy [171]. One of the most important consequences is the dysfunction of Na+/Ca2+ exchange regulation [172]. Mutations in the PINK1 gene are the most common cause of recessive familial PD [170]. Gandhi et al. demonstrated that kinase-dead mutant K219M-PINK1 promotes a dysfunction of Na+/Ca2+ exchanger with the consequent accumulation of Ca2+ in the mitochondria [172]. Calcium overload in the mitochondria stimulates NAPDH oxidase and the production of ROS. The disruption of the electron chain also causes a decrease in glucose transporters in the membrane of neurons by decreasing cellular metabolism. Different studies have shown that the accumulation of ROS stimulates the activity of nSMase2 and the production of Cer [173,174]. Interestingly, the elevation of Cer levels can also take place in the mitochondria by the action of ASAH1 and ASAH2. Moreover, Cer was described to stimulate mitophagy by a mechanism that involves the recruitment of the PINK1 / Parkin complex to the mitochondrial membrane [175].
As mentioned previously, PP2A is involved in the regulation of mitochondrial autophagy. In mouse striatum neurons infected with PINK1 gene silencing lentivirus vectors (LVs) an increase in the phosphorylation of PP2A was observed at residue tyrosine 307 (Y307). This phosphorylation resulted in an inactive form of PP2A in dopaminergic cells and striatum tissues, which indicates the involvement of PP2A in the maintenance of mitochondrial homeostasis by PINK1 [176]. Hannun's group has already shown that the administration of exogenous short-chain Cer (C2-Cer) stimulates the activity of PP2A [177]. In the mentioned work, the authors described that the incubation of PINK1-silenced in differentiated dopaminergic MN9D cells with C2-Cer activated PP2A and thus reduced autophagy levels in a B-cell lymphoma 2 (Bcl-2)-dependent mechanism, a protein implicated in mitochondrial permeabilization and apoptotic processes [176].
3.1.5 SCARB2
SCARB2 gene encodes to lysosomal integral membrane protein 2 (LIMP2), a protein responsible for transporting β-GCase from the ER to the lysosome through interaction with mannose-6-phosphate receptor [93,178]. Different genetic studies have determined the implication of mutations in this gene in the development of PD [179–183]. Moreover, overexpression of α-syn in ASOTg/Tg mouse models showed reduced levels of mannose-6-phosphate receptor type I (also known as MPR300) [184]. Interestingly, Rothaug et al. demonstrated that the loss of LIMP-2 causes a reduction in β-GCase activity that results in a build-up of α-syn and thus neurotoxicity in dopaminergic neurons [185].
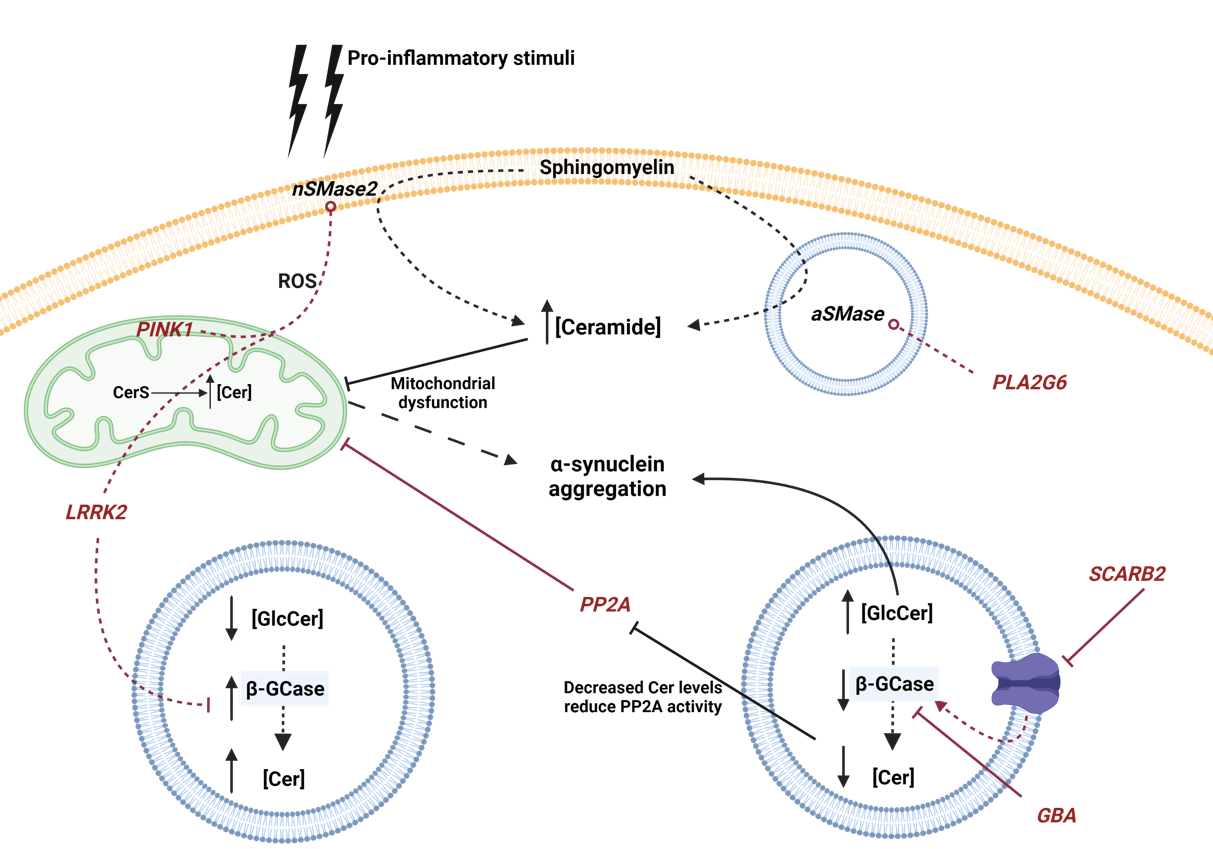
Figure 2. Schematic representation of the influence of genetic mutations involved in PD with Cer metabolism. Genes are represented in italics and red color. Red lines circled at the end indicate increased activity, while a traversed lines at the end indicate inhibition or dysfunction. Neutral Sphingomyelinase 2 (nSMase2), acid sphingomyelinase (aSMase), Ceramide Synthase (CerS), acid β-glucosidase (β-GCase), Ceramide (Cer), GlucosylCeramide (GlcCer) and reactive oxygen species (ROS) are represented by their acronyms.
3.2 ENVIROMENTAL RISKS
Different neurotoxic agents have been described to affect dopaminergic neurons. In this section, a brief summary of some of these agents will be made, and their relationship with Cer metabolism will be addressed.
MPTP is a chemical compound able to cross the blood-brain-barrier (BBB). Once in the brain, monoamine oxidase B enzyme (MAOB) catalyses MPTP to produce 1-methyl-4-phenylpyridinium ion (MPP+), a toxic metabolite that accumulate in dopaminergic neurons and inhibits mitochondrial complex I [186]. As a consequence, there is a reduction in ATP levels and generation of free radicals. Furthermore, MPP+ increase vesicular dopamine in cytoplasm, where it can also produce ROS [187]. However, this PD model does not present Lewy bodies, one of the characteristic hallmarks of PD. Meanwhile, Rotenone is a lipophilic pesticide found in some tropical plants. Due to its chemical characteristics, it can cross the BBB and internalize in dopaminergic neurons. Like MPP+, Rotenone blocks mitochondrial complex I and stimulates ROS production. In addition, Rotenone also inhibits antioxidant enzymes, such as superoxide dismutase (SOD), catalase or glutathione (GSH). This increase in ROS and the inhibition of enzymes involved in their reduction causes degeneration of dopaminergic neurons in the substantia nigra with develop of Lewy Bodies. Moreover, it has also been reported that Rotenone induces inflammation, depolarization of microtubules and inhibition of autophagy [188].
In the hippocampus of PD mouse models induced by MPTP was observed that nSMase was down-regulated [189]. Previously, 1,25-dihydroxyvitamin D3 was described as activator of SMase activity [190]. In vitro studies in hippocampal PD mice neurons induced by MPTP with consequent upregulation of iNOS and downregulation of nSMase, showed a reduced levels of SM by upregulation of nSMase by 1,25-dihydroxyvitamin D3, related with an improve in synaptic plasticity [189]. Interestingly, treatment with MPP+ showed a significant alteration in SphKs and S1P lyase expression in SH-SY5Y dopaminergic neuronal cells [191,192].
Several reports have shown evidence suggesting a critical role of the toll like receptor 4 (TLR4) in inflammatory responses and neuronal death [193–196]. Both the microglia- mediated inflammatory pathway and aggregated α-syn activate different mechanisms to stimulate inflammation and contribute to neurodegenerative progression in PD via upregulation of TLR2 and TLR4 [197]. Recent studies in TLR4 knockout mice showed protection against MPTP toxicity, with attenuation of motor deficits and a reduction in α-syn dysfunction and neuroinflammation [193]. Interestingly, another study with these mice described an increase in the expression of nSMase that led to a decrease in SM levels and an increase in Cer, which causes a higher sensitivity to MPTP [198].
Rotenone has been described to inhibit PP2A, preventing the dephosphorylating of α-syn and, in turn increasing its aggregation [199]. Furthermore, C2-Cer activates PP2A and counteracted the neurotoxic effects derived from Rotenone treatment and decrease α-syn aggregation [199]. Additionally, in erythrocytes that are known to lack mitochondria, treatment with Rotenone stimulates apoptosis due to an increase in Cer levels, by a mitochondrial-independent mechanism [200]. As showed above, Rotenone inhibits also GSH. Studies in hepatocytes demonstrated that the decrease in GSH activity was correlated with increases in the activity of nSMase2, and this was related to an IL-1β hyperresponsiveness, increasing the pro-inflammatory response [201]. Interestingly, recent study has demonstrated that ASAH1 overexpression increase GSH levels and reduce oxidative stress in fibroblasts derived from Niemann-Pick’s disease type C1 [97].
4. Human Sphingolipidomics of Ceramide Metabolism in PD
In recent years, studies on sphingolipid levels by mass spectrometry (sphingolipidomics) have gained importance in different fields of medicine and scientific research [202] (table 1). It has been reported that sphingolipids metabolism is related to the progression of different carcinogenic processes, as well as resistance to anti-cancer treatments [203–205]. Therefore, the study of the evolution of the different species will allow a more effective diagnosis and treatment. Regarding PD, differences in the levels of Cer, SM and GM1, among other sphingolipids, have been described in post-mortem brain samples, blood and CSF from patients with different neuronal degeneration [17,23,206].
PD post-mortem brain tissue from anterior cingulate cortex, showed a drastic reduction in long-chain Cer (such as C24:1 and C24:0-Cer) levels. Interestingly, an upregulation of CerS1 and CerS4 was observed, which could be due to a compensation system to increase certain Cer species [207][207]Meanwhile, comparing plasma samples from patients with GBA mutations and non-carrier patients, an increase in Cer and LacCer levels were described, in addition to a decrease in C1P levels [169]. Additionally, in a recent study of post-mortem CSF samples have been described an increase in Cer and SM levels, which correlated with neuropathological staging and disease duration, a fact that further supports the ability of sphingolipids as biomarkers [208]. Moreover, plasma C14:0 and C24:1-Cer levels were significantly higher in PD with dementia than in PD with no cognitive impairment and normal controls. Interestingly, C22:0, C20:0 and C18:0-Cer were associated with hallucinations, anxiety and disturbances of sleep behaviour, respectively [209]. Along with these observations, also plasma Cer (C16:0, C18:0, C20:0, C22:0 and C24:1) species were found elevated in PD non-GBA mutation carriers, with higher levels associated with worse cognitive function [210].
A general increase of long chain SM and Cer levels was also detected in the primary visual cortex [211], as well as, an increase of SM levels in the substantia nigra but only in male PD patients [212]. However, no changes in sphingolipids were observed in the putamen or cerebellum of GBA PD patients [213]. Nonetheless, one study reported reduced plasma levels of SM (d30:1, 32:1 and 39:1) in early PD patients compared to controls [214].
Some GlcCer species, including C18:0, C 20:0, C 22:0, C 24:1, and C 24:0 GlcCer in brain tissue revealed a correlation between increased levels of these metabolites and PD severity [215]. In addition, plasma GlcCer (C16:0, C22:0 and C24:0) species were increased in PD non-GBA mutation carriers, and were correlated with greater cognitive impairment [210]. In contrast, GlcCer C24:1 was selectively decreased in PD patients with a reduction of C18:1-Cer in frontal cortex [216]. Moreover, no significant differences have been found between the different PD groups and controls in CSF samples [217].
Gangliosides are complex sphingolipids involved in the regulation of inflammatory processes. Their involvement in PD develop has been observed in several studies, although the mechanisms are still uncertain [218]. GM1 can increase the internalization process of α-syn by the glia, increasing α-syn clearance [219] and inhibit the aggregation [220]. By contrast, GM3 accelerates α-syn aggregation by a mechanism dependent on the biophysical conditions of both molecules [221].
Ganglioside GM1 has been reported to be decreased in dopaminergic neurons in substantia nigra of PD patients versus age-matched controls [222]. In plasma from PD patients with GBA mutations and/or LRRK2 G2019S mutations, increased levels of ganglioside-NANA-3 (a precursor of complex sphingolipids) were observed, possibly due to a reduction in the activity of β-GCase [223]. In addition, in non-carriers of GBA or LRRK2-G2019S mutations PD patients GM3 species (d18:1/24:1 and d18:1/26:0) were found elevated in comparation with controls, correlated with high levels of GlcCer species in plasma [224].
|
Sphingolipid species |
Levels |
Source |
Type of PD |
Notes |
Ref. |
|
Cer |
↑ |
Serum |
GBA mutation |
Possible early development of PD and worsening of symptoms |
[169] |
|
↑ |
Post-mortem CSF |
Not specified |
Correlation with neuropathological staging and disease duration |
[208] |
|
|
↑ |
Primary visual cortex |
Sporadic |
Contribution to neuronal dysfunction |
[211] |
|
|
Long-chain Cer (such as C24:1 and C24:0-Cer) |
↓ |
Anterior cingulate cortex |
Impartment of salvage pathway by increased CerS1 expression |
[207] |
|
|
C14:0-Cer |
↑ |
Plasma |
PD with dementia |
Association with delayed free recall and cognition |
[225] |
|
C16:0-Cer |
↑ |
Plasma |
Non-GBA mutation |
Association with worse cognition |
[210] |
|
C18:0-Cer |
↑ |
Plasma |
Non-GBA mutation |
Association with worse cognition |
[210] |
|
↑* |
PD with dementia |
Association with sleep behaviour disturbance |
[225] |
||
|
C18:1-Cer |
↓ |
Frontal cortex |
Not specified |
Increased formation of diacylglycerols (DAGs) |
[216] |
|
C20:0-Cer |
↑* |
Plasma |
PD with dementia |
Association with anxiety |
[225] |
|
↑ |
Non-GBA mutation |
Association with worse cognition |
[210] |
||
|
C22:0-Cer |
↑ |
Plasma |
Non-GBA mutation |
Association with worse cognition |
[210] |
|
↑* |
PD with dementia |
Association with hallucination |
[225] |
||
|
C24:1-Cer |
↑ |
Plasma |
PD with dementia |
Association with the score of immediate verbal recall and delayed free recall |
[225] |
|
↑ |
Non-GBA mutation |
Association with worse cognition |
[210] |
||
|
GlcCer |
↑ |
Plasma |
Non-GBA or Non-LRRK2 G2019S mutation |
Association with worse cognition |
[210,224] |
|
GlcCer C16:0 |
↑ |
Plasma |
Non-GBA mutation |
Association with worse cognition |
[210] |
|
GlcCer C18:0 |
↑* |
Temporal cortex |
Not specified |
Correlation with PD severity |
[215] |
|
GlcCer C20:0 |
↑* |
Temporal cortex |
Not specified |
Correlation with PD severity |
[215] |
|
GlcCer C22:0 |
↑* |
Temporal cortex |
Not specified |
Correlation with PD severity |
[215] |
|
↑ |
Plasma |
Non-GBA mutation |
Tendency to association with worse cognition |
[210] |
|
|
GlcCer C24:0 |
↑ |
Plasma |
Non-GBA mutation |
Association with worse cognition |
[210] |
|
↑* |
Temporal cortex |
Not specified |
Correlation with PD severity |
[215] |
|
|
GlcCer C24:1 |
↑* |
Temporal cortex |
Not specified |
Correlation with PD severity |
[215] |
|
↓ |
Frontal cortex |
Not specified |
Increased formation of DAGs |
[216] |
|
|
LacCer |
↑ |
Serum |
GBA mutation |
Proposed as novel biomarker for increased risk of PD develop |
[169] |
|
C1P |
↓ |
Serum |
GBA mutation |
Proposed as novel biomarker for increased risk of PD develop |
[169] |
|
SM |
↑ |
Post-mortem CSF |
Not specified |
Correlation with neuropathological staging and disease duration |
[211] |
|
↑ |
Substantia nigra |
Male PD |
Caused by enrichment in Lewy Bodies |
[212] |
|
|
Long-chain SM |
↑ |
Primary visual cortex |
Sporadic |
Contribution to neuronal dysfunction |
[211] |
|
SM d30:1 |
↓* |
Plasma |
Not specified |
Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination |
[214] |
|
SM d32:1 |
↓* |
Plasma |
Not specified |
Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination |
[214] |
|
SM d39:1 |
↓* |
Plasma |
Not specified |
Due to dysregulation of sphingolipids metabolism. Possibly involved in demyelination |
[214] |
|
GM1 |
↓ |
Substantia nigra dopaminergic neurons |
Sporadic |
Loss of neuroprotection and acceleration of α-syn formation |
[222] |
|
Ganglioside-NANA-3 |
↑ |
Plasma |
GBA or/and LRRK2 G2019S mutation |
Acceleration of α-syn formation |
[223] |
|
GM3 d18:1/24:1 |
↑ |
Plasma |
Non-GBA or Non-LRRK2 G2019S mutation |
Acceleration of α-syn formation |
[224] |
|
GM3 d18:1/26:0 |
↑ |
Plasma |
Non-GBA or Non-LRRK2 G2019S mutation |
Acceleration of α-syn formation |
[224] |
Table 1. Summary table of human sphingolipidomics. The up arrows refer to an increase in levels compared to the controls, while the down arrows refer to a reduction. The asterisk indicates that the data were not statistically significant. Ceramide (Cer), ceramide 1-phosphate (C1P), sphingomyelin (SM), glucosyl-ceramide (GlcCer) and lactosyl-ceramide (LacCer).
5. Concluding remarks
The dual function of sphingolipids as structural molecules and second messengers makes them vitally important molecules for the maintenance of cell and tissue homeostasis. It has been demonstrated that alterations of sphingolipid metabolism are related to PD progression, and its aggressiveness by a large number of studies carried out in different animal models, in vitro studies and human samples. However, the molecular mechanisms that give rise to this alteration remain incompletely understood. Studies in which the therapeutic targets are the enzymes involved in the metabolism of sphingolipids are of vital importance.
In recent years, sphingolipidomic studies in patient samples have emerged as a new tool to evaluate the aggressiveness and establish specific treatments of certain diseases, including PD. Moreover, sphingolipid levels in PD patients with different symptoms and progress have demonstrated the viability of sphingolipidomics for both diagnosis and specific treatments. These data support the potential of sphingolipids as biomarkers of PD. Furthermore, transcriptional studies have shown variations in the expression of different enzymes involved in sphingolipid metabolism. However, only a few studies have been conducted to determine their involvement in neurological impairment in PD.
Future experiments that aim to understand the involvement of sphingolipids in the development of PD will be of vital importance for the development of therapies and an early diagnosis.
Author Contributions: Conceptualization, A.O.; writing—original draft preparation, A.C., M.A-N., A.G-M. and A.O.; writing—review and editing, all the authors.; visualization, A.C. and C.C-P.; supervision, A.O.; project administration, T.S.; funding acquisition, A.G.M. and T.S. All authors have read and agreed to the published version of the manuscript.
Funding: This study was partially supported by grants from the Xunta de Galicia (Consellería de Economía e Industria: IN607A2018/3 & IN607D 2020/09), and Science Ministry of Spain (RTI2018-102165-B-I00 & RTC2019-007373-1). Furthermore, this study was also supported by grants from the INTERREG Atlantic Area (EAPA_791/2018_ NEUROATLANTIC project), INTERREG V A España Portugal (POCTEP) (0624_2IQBIONEURO_6_E), and the European Regional Development Fund (ERDF). Work in AGM lab is supported by grant IT-1106-16 from “Departamento de Educación, Universidades e Investigación” (Gobierno Vasco, Gasteiz-Virtoria, Spain). Moreover, M. Aramburu-Núñez (IFI18/00008) is recipient of iPFIS contract, and Dr. Sobrino (CPII17/00027) is recipient of a research contract from the Miguel Servet Program from the Instituto de Salud Carlos III. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments: The figure 1 was made with BioRender.com software
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s Disease. Journal of Neural Transmission 2017, 124, 901–905, doi:10.1007/s00702-017-1686-y.
2. Dextera, D.T.; Jenner, P. Parkinson Disease: From Pathology to Molecular Disease Mechanisms. Free Radical Biology and Medicine 2013, 62, 132–144, doi:10.1016/j.freeradbiomed.2013.01.018.
3. Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. The Lancet Neurology 2020, 19, 170–178, doi:10.1016/S1474-4422(19)30287-X.
4. von Campenhausen, S.; Winter, Y.; Rodrigues e Silva, A.; Sampaio, C.; Ruzicka, E.; Barone, P.; Poewe, W.; Guekht, A.; Mateus, C.; Pfeiffer, K.P.; et al. Costs of Illness and Care in Parkinson’s Disease: An Evaluation in Six Countries. European Neuropsychopharmacology 2011, 21, 180–191, doi:10.1016/j.euroneuro.2010.08.002.
5. Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. International Journal of Molecular Sciences 2019, 20, doi:10.3390/ijms20133304.
6. Alessenko, A. v.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Frontiers in Neurology 2020, 11, 1–11, doi:10.3389/fneur.2020.00437.
7. Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends in Endocrinology and Metabolism 2017, 28, 506–518, doi:10.1016/j.tem.2017.03.005.
8. Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays in Biochemistry 2020, 64, 579–589, doi:10.1042/EBC20190091.
9. Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. Journal of Neurochemistry 2021, 156, 403–414, doi:10.1111/jnc.15133.
10. Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. International Journal of Molecular Sciences 2021, 22, 5793, doi:10.3390/ijms22115793.
11. Ouro, A.; Arana, L.; Gangoiti, P.; Gomez-Muñoz, A. Role of Ceramide 1-Phosphate in the Regulation of Cell Survival and Inflammation. Biochemistry 2012, 4, doi:10.5772/32849.
12. Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis 2010, 9, 15, doi:10.1186/1476-511X-9-15.
13. Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; et al. Control of Metabolism and Signaling of Simple Bioactive Sphingolipids: Implications in Disease. Prog Lipid Res 2010, 49, 316–334, doi:10.1016/j.plipres.2010.02.004.
14. Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat Rev Mol Cell Biol 2008, 9, 139–150, doi:10.1038/nrm2329.
15. Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal Fluid Sphingolipids, β-Amyloid, and Tau in Adults at Risk for Alzheimer’s Disease. Neurobiology of Aging 2014, 35, 2486–2494, doi:10.1016/j.neurobiolaging.2014.05.019.
16. Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends in Endocrinology and Metabolism 2019, 30, 106–117, doi:10.1016/j.tem.2018.11.003.
17. van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as Prognostic Biomarkers of Neurodegeneration, Neuroinflammation, and Psychiatric Diseases and Their Emerging Role in Lipidomic Investigation Methods. Advanced Drug Delivery Reviews 2020, 159, 232–244, doi:10.1016/j.addr.2020.04.009.
18. Iqbal, J.; Walsh, M.T.; Hammad, S.M.; Hussain, M.M. Sphingolipids and Lipoproteins in Health and Metabolic Disorders. Trends in Endocrinology and Metabolism 2017, 28, 506–518, doi:10.1016/j.tem.2017.03.005.
19. Gulbins, A.; Grassm, H.; Hoehn, R.; Wilker, B.; Soddemann, M.; Kohnen, M.; Edwards, M.J.; Kornhuber, J.; Gulbins, E. Regulation of Neuronal Stem Cell Proliferation in the Hippocampus by Endothelial Ceramide. Cellular Physiology and Biochemistry 2016, 39, 790–801, doi:10.1159/000447789.
20. Schultz, A.; Larsson, C. Ceramide Influences Neurite Outgrowth and Neuroblastoma Cell Apoptosis Regulated by Novel Protein Kinase C Isoforms. Journal of Neurochemistry 2004, 89, 1427–1435, doi:10.1111/j.1471-4159.2004.02431.x.
21. Cruciani-Guglielmacci, C.; López, M.; Campana, M.; le Stunff, H. Brain Ceramide Metabolism in the Control of Energy Balance. Frontiers in Physiology 2017, 8, 787, doi:10.3389/fphys.2017.00787.
22. Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and Neurodegeneration: Susceptibility of Neurons and Oligodendrocytes to Cell Damage and Death. Journal of the Neurological Sciences 2009, 278, 5–15, doi:10.1016/j.jns.2008.12.010.
23. Pujol-Lereis, L.M. Alteration of Sphingolipids in Biofluids: Implications for Neurodegenerative Diseases. International Journal of Molecular Sciences 2019, 20, 3564–3584, doi:10.3390/ijms20143564.
24. Platt, F.M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75.
25. Watters, R.J.; Kester, M.; Tran, M.A.; Loughran, T.P.; Liu, X. Development and Use of Ceramide Nanoliposomes in Cancer. Methods in enzymology 2012, 508, 89–108, doi:10.1016/B978-0-12-391860-4.00005-7.
26. Gomez-Muñoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordoñez, M. Control of Inflammatory Responses by Ceramide, Sphingosine 1-Phosphate and Ceramide 1-Phosphate. Progress in Lipid Research 2016, doi:10.1016/j.plipres.2015.09.002.
27. Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Advances in experimental medicine and biology 2019, 1161, 169–191, doi:10.1007/978-3-030-21735-8_15.
28. Wattenberg, B.W. Kicking off Sphingolipid Biosynthesis: Structures of the Serine Palmitoyltransferase Complex. Nature Structural and Molecular Biology 2021, 28, 229–231, doi:10.1038/s41594-021-00562-0.
29. Kim, J.L.; Mestre, B.; Shin, S.-H.; Futerman, A.H. Ceramide Synthases: Reflections on the Impact of Dr. Lina M. Obeid. Cellular Signalling 2021, 109958, doi:10.1016/j.cellsig.2021.109958.
30. Mignard, V.; Dubois, N.; Lanoé, D.; Joalland, M.P.; Oliver, L.; Pecqueur, C.; Heymann, D.; Paris, F.; Vallette, F.M.; Lalier, L. Sphingolipids Distribution at Mitochondria-Associated Membranes (MAM) upon Induction of Apoptosis. Journal of Lipid Research 2020, 61, 1025–1037, doi:10.1194/JLR.RA120000628.
31. Novgorodov, S.A.; Gudz, T.I. Ceramide and Mitochondria in Ischemic Brain Injury. International Journal of Biochemistry and Molecular Biology 2011, 2, 347–361.
32. Yu, J.; Novgorodov, S.A.; Chudakova, D.; Zhu, H.; Bielawska, A.; Bielawski, J.; Obeid, L.M.; Kindy, M.S.; Gudz, T.I. JNK3 Signaling Pathway Activates Ceramide Synthase Leading to Mitochondrial Dysfunction. Journal of Biological Chemistry 2007, 282, 25940–25949, doi:10.1074/jbc.M701812200.
33. Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Rufus Sweeney, C.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392, doi:10.1126/science.aav3722.
34. Ohi, K.; Ursini, G.; Li, M.; Shin, J.H.; Ye, T.; Chen, Q.; Tao, R.; Kleinman, J.E.; Hyde, T.M.; Hashimoto, R.; et al. DEGS2 Polymorphism Associated with Cognition in Schizophrenia Is Associated with Gene Expression in Brain. Translational Psychiatry 2015, 5, 550, doi:10.1038/tp.2015.45.
35. Karsai, G.; Kraft, F.; Haag, N.; Christoph Korenke, G.; Hänisch, B.; Othman, A.; Suriyanarayanan, S.; Steiner, R.; Knopp, C.; Mull, M.; et al. DEGS1-Associated Aberrant Sphingolipid Metabolism Impairs Nervous System Function in Humans. Journal of Clinical Investigation 2019, 129, 1229–1239, doi:10.1172/JCI124159.
36. Goi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Letters 2002, 531, 38–46, doi:10.1016/S0014-5793(02)03482-8.
37. Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal Structure of Mammalian Acid Sphingomyelinase. Nature Communications 2016, 7, 1–9, doi:10.1038/ncomms12196.
38. Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, N.; Hannun, Y.A. The Extended Family of Neutral Sphingomyelinases. Biochemistry 2006, 45, 11247–11256, doi:10.1021/bi061307z.
39. Goni, F.M.; Alonso, A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett 2002, 531, 38–46.
40. Cataldi, S.; Borrelli, A.; Ceccarini, M.R.; Nakashidze, I.; Codini, M.; Belov, O.; Ivanov, A.; Krasavin, E.; Ferri, I.; Conte, C.; et al. Acid and Neutral Sphingomyelinase Behavior in Radiation-Induced Liver Pyroptosis and in the Protective/Preventive Role of RMnSOD. International Journal of Molecular Sciences 2020, 21, doi:10.3390/ijms21093281.
41. Kornhuber, J.; Rhein, C.; Müller, C.P.; Mühle, C. Secretory Sphingomyelinase in Health and Disease. Biological Chemistry 2015, 396, 707–736.
42. Jenkins, R.W.; Canals, D.; Idkowiak-Baldys, J.; Simbari, F.; Roddy, P.; Perry, D.M.; Kitatani, K.; Luberto, C.; Hannun, Y.A. Regulated Secretion of Acid Sphingomyelinase: Implications for Selectivity of Ceramide Formation. J Biol Chem 2010, 285, 35706–35718.
43. Becker, K.A.; Riethmuller, J.; Luth, A.; Doring, G.; Kleuser, B.; Gulbins, E. Acid Sphingomyelinase Inhibitors Normalize Pulmonary Ceramide and Inflammation in Cystic Fibrosis. Am J Respir Cell Mol Biol 2010, 42, 716–724, doi:10.1165/rcmb.2009-0174OC.
44. Gomez-Muñoz, A.; Gangoiti, P.; Arana, L.; Ouro, A.; Rivera, I.G.; Ordoñez, M.; Trueba, M. New Insights on the Role of Ceramide 1-Phosphate in Inflammation. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 2013, 1831, 1060–1066, doi:10.1016/j.bbalip.2013.02.001.
45. Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiology of Disease 23, 178–189, doi:10.1016/j.nbd.2006.02.010.
46. Morad, S.A.F.; Cabot, M.C. Ceramide-Orchestrated Signalling in Cancer Cells. Nat Rev Cancer 2012, 13, 51–65, doi:10.1038/nrc3398.
47. Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid Sphingomyelinase Deficient Mice: A Model of Types A and B Niemann–Pick Disease. Nature Genetics 1995, 10, 288–293, doi:10.1038/ng0795-288.
48. Schuchman, E.H.; Wasserstein, M.P. Types A and B Niemann-Pick Disease. Best Practice and Research: Clinical Endocrinology and Metabolism 2015, 29, 237–247, doi:10.1016/j.beem.2014.10.002.
49. Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. NeuroMolecular Medicine 2010, 12, 320–330, doi:10.1007/s12017-010-8120-z.
50. Shamseddine, A.A.; Airola, M. v.; Hannun, Y.A. Roles and Regulation of Neutral Sphingomyelinase-2 in Cellular and Pathological Processes. Advances in Biological Regulation 2015, 57, 24–41, doi:10.1016/j.jbior.2014.10.002.
51. Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and Characterization of the Mammalian Brain-Specific, Mg2+-Dependent Neutral Sphingomyelinase. Proceedings of the National Academy of Sciences of the United States of America 2000, 97, 5895–5900, doi:10.1073/pnas.97.11.5895.
52. Cataldi, S.; Arcuri, C.; Hunot, S.; Légeron, F.P.; Mecca, C.; Garcia-Gil, M.; Lazzarini, A.; Codini, M.; Beccari, T.; Tasegian, A.; et al. Neutral Sphingomyelinase Behaviour in Hippocampus Neuroinflammation of MPTP-Induced Mouse Model of Parkinson’s Disease and in Embryonic Hippocampal Cells. Mediators of Inflammation 2017, 2017, doi:10.1155/2017/2470950.
53. Tabatadze, N.; Savonenko, A.; Song, H.; Bandaru, V.V.R.; Chu, M.; Haughey, N.J. Inhibition of Neutral Sphingomyelinase-2 Perturbs Brain Sphingolipid Balance and Spatial Memory in Mice. Journal of Neuroscience Research 2010, 88, 2940–2951, doi:10.1002/jnr.22438.
54. Gu, L.Z.; Huang, B.S.; Shen, W.; Gao, L.; Ding, Z.Z.; Wu, H.W.; Guo, J. Early Activation of NSMase2/Ceramide Pathway in Astrocytes Is Involved in Ischemia-Associated Neuronal Damage via Inflammation in Rat Hippocampi. Journal of Neuroinflammation 2013, 10, 1–16, doi:10.1186/1742-2094-10-109.
55. Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Human Mutation 2008, 29, 567–583, doi:10.1002/humu.20676.
56. Velayati, A.; Haung Yu, W.; Sidransky, E. The Role of Glucocerebrosidase Mutations in Parkinson Disease and Lewy Body Disorders. Current Neurology and Neuroscience Reports 2010, 10, 190–198, doi:10.1007/s11910-010-0102-x.
57. Coant, N.; Hannun, Y.A. Neutral Ceramidase: Advances in Mechanisms, Cell Regulation, and Roles in Cancer. Adv Biol Regul 2019, 71, 141–146, doi:10.1016/j.jbior.2018.10.005.
58. Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/Alkaline and Acid Ceramidase Activities Are Actively Released by Murine Endothelial Cells. Biochem Biophys Res Commun 2000, 275, 746–751, doi:10.1006/bbrc.2000.3370.
59. Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gomez-Muñoz, A.; Gomez-Munoz, A. Activation of Protein Kinase C-Alpha Is Essential for Stimulation of Cell Proliferation by Ceramide 1-Phosphate. FEBS Lett 2010, 584, 517–524, doi:S0014-5793(09)01025-4 [pii] 10.1016/j.febslet.2009.11.086.
60. Gangoiti, P.; Bernacchioni, C.; Donati, C.; Cencetti, F.; Ouro, A.; Gómez-Muñoz, A.; Bruni, P.; Gomez-Munoz, A.; Bruni, P. Ceramide 1-Phosphate Stimulates Proliferation of C2C12 Myoblasts. Biochimie 2012, 94, 597–607, doi:10.1016/j.biochi.2011.09.009.
61. Ouro, A.; Arana, L.; Gangoiti, P.; Rivera, I.G.; Ordoñez, M.; Trueba, M.; Lankalapalli, R.S.; Bittman, R.; Gomez-Muñoz, A. Ceramide 1-Phosphate Stimulates Glucose Uptake in Macrophages. Cellular Signalling 2013, 25, 786–795, doi:10.1016/j.cellsig.2013.01.009.
62. Gangoiti, P.; Granado, M.H.; Wei, S.; Kong, J.Y.; Steinbrecher, U.P.; Gómez-muñoz, A. Ceramide 1-Phosphate Stimulates Macrophage Proliferation through Activation of the PI3-Kinase / PKB , JNK and ERK1 / 2 Pathways. 2008, 20, 726–736, doi:10.1016/j.cellsig.2007.12.008.
63. Ouro, A.; Arana, L.; Riazy, M.; Zhang, P.; Gomez-Larrauri, A.; Steinbrecher, U.; Duronio, V.; Gomez-Muñoz, A. Vascular Endothelial Growth Factor Mediates Ceramide 1-Phosphate-Stimulated Macrophage Proliferation. Experimental Cell Research 2017, 361, 277–283, doi:10.1016/j.yexcr.2017.10.027.
64. Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A.; Gómez-Muñoz, A. Involvement of Nitric Oxide in the Promotion of Cell Survival by Ceramide 1-Phosphate. FEBS Lett 2008, 582, 2263–2269, doi:10.1016/j.febslet.2008.05.027.
65. Gomez-Munoz, A.; Kong, J.; Salh, B.; Steinbrecher, U.P. Sphingosine-1-Phosphate Inhibits Acid Sphingomyelinase and Blocks Apoptosis in Macrophages. FEBS Lett 2003, 539, 56–60.
66. Newcomb, B.; Rhein, C.; Mileva, I.; Ahmad, R.; Clarke, C.J.; Snider, J.; Obeid, L.M.; Hannun, Y.A. Identification of an Acid Sphingomyelinase Ceramide Kinase Pathway in the Regulation of the Chemokine CCL5. Journal of Lipid Research 2018, 59, 1219–1229, doi:10.1194/jlr.M084202.
67. Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gómez-Muñoz, A. Ceramide 1-Phosphate Inhibits Serine Palmitoyltransferase and Blocks Apoptosis in Alveolar Macrophages. Biochim Biophys Acta 2009, 1791, 263–272, doi:10.1016/j.bbalip.2009.01.023.
68. Gomez-Munoz, A.; Kong, J.Y.; Parhar, K.; Wang, S.W.; Gangoiti, P.; Gonzalez, M.; Eivemark, S.; Salh, B.; Duronio, V.; Steinbrecher, U.P. Ceramide-1-Phosphate Promotes Cell Survival through Activation of the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. FEBS Lett 2005, 579, 3744–3750, doi:10.1016/j.febslet.2005.05.067.
69. Mishra, S.K.; Gao, Y.G.; Deng, Y.; Chalfant, C.E.; Hinchcliffe, E.H.; Brown, R.E. CPTP: A Sphingolipid Transfer Protein That Regulates Autophagy and Inflammasome Activation†. Autophagy 2018, 14, 862–879, doi:10.1080/15548627.2017.1393129.
70. Goetzl, E.J.; Wang, W.; McGiffert, C.; Huang, M.C.; Graler, M.H. Sphingosine 1-Phosphate and Its G Protein-Coupled Receptors Constitute a Multifunctional Immunoregulatory System. J Cell Biochem 2004, 92, 1104–1114.
71. Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. NeuroMolecular Medicine 2021, 23, 211–223, doi:10.1007/s12017-020-08614-2.
72. Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-Phosphate Stimulates Proliferation and Migration of Satellite Cells: Role of S1P Receptors. Biochim Biophys Acta 2012, 1823, 439–450, doi:10.1016/j.bbamcr.2011.11.016.
73. Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial Sphingosine 1-Phosphate Receptors Promote Vascular Normalization and Antitumor Therapy. Proceedings of the National Academy of Sciences of the United States of America 2020, 117, 3157–3166, doi:10.1073/pnas.1906246117.
74. Saba, J.D. Fifty Years of Lyase and a Moment of Truth: Sphingosine Phosphate Lyase from Discovery to Disease. Journal of Lipid Research 2019, 60, 456–463, doi:10.1194/jlr.S091181.
75. Mitroi, D.N.; Karunakaran, I.; Gräler, M.; Saba, J.D.; Ehninger, D.; Ledesma, M.D.; van Echten-Deckert, G. SGPL1 (Sphingosine Phosphate Lyase 1) Modulates Neuronal Autophagy via Phosphatidylethanolamine Production. Autophagy 2017, 13, 885–899, doi:10.1080/15548627.2017.1291471.
76. Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathologica 2009, 118, 5–36.
77. Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Medical Clinics of North America 103, 263–293.
78. Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B. Serum Ceramides Increase the Risk of Alzheimer Disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641, doi:10.1212/WNL.0b013e318264e380.
79. Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H. v; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. Journal of Alzheimer’s Disease 2012, 29, 537–547, doi:10.3233/JAD-2011-111202.
80. Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Molecular Neurobiology 2019, 56, 5436–5455, doi:10.1007/s12035-018-1448-3.
81. Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and Sphingomyelinases in Senile Plaques. Neurobiology of Disease 2014, 65, 193–201, doi:10.1016/j.nbd.2014.01.010.
82. Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H. V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. Journal of Alzheimer’s Disease 2012, 29, 537–547, doi:10.3233/JAD-2011-111202.
83. Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide Stabilizes β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid β-Peptide Biogenesis. Journal of Biological Chemistry 2003, 278, 19777–19783, doi:10.1074/jbc.M300466200.
84. Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiology of Disease 2006, 23, 178–189, doi:10.1016/j.nbd.2006.02.010.
85. Desbène, C.; Malaplate-Armand, C.; Youssef, I.; Garcia, P.; Stenger, C.; Sauvée, M.; Fischer, N.; Rimet, D.; Koziel, V.; Escanyé, M.C.; et al. Critical Role of CPLA2 in Aβ Oligomer-Induced Neurodegeneration and Memory Deficit. Neurobiology of Aging 2012, 33, 1123.e17-1123.e29, doi:10.1016/j.neurobiolaging.2011.11.008.
86. Jana, A.; Pahan, K. Fibrillar Amyloid-β-Activated Human Astroglia Kill Primary Human Neurons via Neutral Sphingomyelinase: Implications for Alzheimer’s Disease. Journal of Neuroscience 2010, 30, 12676–12689, doi:10.1523/JNEUROSCI.1243-10.2010.
87. Lee, J.T.; Xu, J.; Lee, J.M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β Peptide Induces Oligodendrocyte Death by Activating the Neutral Sphingomyelinase-Ceramide Pathway. Journal of Cell Biology 2004, 164, 123–131, doi:10.1083/jcb.200307017.
88. Yang, D.I.; Yeh, C.H.; Chen, S.; Xu, J.; Hsu, C.Y. Neutral Sphingomyelinase Activation in Endothelial and Glial Cell Death Induced by Amyloid Beta-Peptide. Neurobiology of Disease 2004, 17, 99–107, doi:10.1016/j.nbd.2004.06.001.
89. Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. Journal of Neuroscience 2011, 31, 6850–6857, doi:10.1523/JNEUROSCI.6467-10.2011.
90. Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced Sphingosine Kinase-1 and Enhanced Sphingosine 1-Phosphate Lyase Expression Demonstrate Deregulated Sphingosine 1-Phosphate Signaling in Alzheimer’s Disease. Acta Neuropathologica Communications 2014, 2, doi:10.1186/2051-5960-2-12.
91. Platt, F.M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75, doi:10.1038/nature13476.
92. Paciotti, S.; Albi, E.; Parnetti, L.; Beccari, T. Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease. Journal of Clinical Medicine 2020, 9, 594, doi:10.3390/jcm9020594.
93. Zhao, Y.; Ren, J.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Lysosome Sorting of β-Glucocerebrosidase by LIMP-2 Is Targeted by the Mannose 6-Phosphate Receptor. Nature Communications 2014, 5, doi:10.1038/ncomms5321.
94. Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. A Rare Lysosomal Enzyme Gene SMPD1 Variant (p.R591C) Associates with Parkinson’s Disease. Neurobiology of Aging 34, 2890 13–2890 15, doi:10.1016/j.neurobiolaging.2013.06.010.
95. Conte, C.; Arcuri, C.; Cataldi, S.; Mecca, C.; Codini, M.; Ceccarini, M.R.; Patria, F.F.; Beccari, T.; Albi, E. Niemann-Pick Type a Disease: Behavior of Neutral Sphingomyelinase and Vitamin D Receptor. International Journal of Molecular Sciences 2019, 20, doi:10.3390/ijms20092365.
96. Vanier, M.T. Niemann-Pick diseases. In Handbook of Clinical Neurology; Elsevier B.V., 2013; Vol. 113, pp. 1717–1721.
97. Torres, S.; Solsona-Vilarrasa, E.; Nuñez, S.; Matías, N.; Insausti-Urkia, N.; Castro, F.; Casasempere, M.; Fabriás, G.; Casas, J.; Enrich, C.; et al. Acid Ceramidase Improves Mitochondrial Function and Oxidative Stress in Niemann-Pick Type C Disease by Repressing STARD1 Expression and Mitochondrial Cholesterol Accumulation. Redox Biology 2021, 102052, doi:10.1016/j.redox.2021.102052.
98. Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine Accumulation in Tissues from Patients with Gaucher Disease: Correlation with Phenotype and Genotype. Molecular Genetics and Metabolism 76, 262–270, doi:10.1016/S1096-7192(02)00117-8.
99. Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid Ceramidase Deficiency: Farber Disease and SMA-PME. Orphanet Journal of Rare Diseases 2018, 13, 1–19, doi:10.1186/s13023-018-0845-z.
100. Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as Highly Sensitive Biomarker for the Diagnosis of Farber Disease., doi:10.1038/s41598-017-06604-2.
101. Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922, doi:10.1111/tra.12404.
102. Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s Disease: The Potential Contribution of Galactosylceramidase Deficiency to Neuronal Vulnerability in Late-Onset Synucleinopathies. Journal of Neuroscience Research 2016, 94, 1328–1332, doi:10.1002/jnr.23751.
103. Maglione, V.; Marchi, P.; di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired Ganglioside Metabolism in Huntington’s Disease and Neuroprotective Role of GM1. Journal of Neuroscience 2010, 30, 4072–4080, doi:10.1523/JNEUROSCI.6348-09.2010.
104. Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; et al. Disease‐modifying Effects of Ganglioside GM1 in Huntington’s Disease Models. EMBO Molecular Medicine 2017, 9, 1537–1557, doi:10.15252/emmm.201707763.
105. Pardo, A. di; Basit, A.; Armirotti, A.; Amico, E.; Castaldo, S.; Pepe, G.; Marracino, F.; Buttari, F.; Digilio, A.F.; Maglione, V. De Novo Synthesis of Sphingolipids Is Defective in Experimental Models of Huntington’s Disease. Frontiers in Neuroscience 2017, 11, doi:10.3389/fnins.2017.00698.
106. Yamout, B.I.; Alroughani, R. Multiple Sclerosis. Seminars in Neurology 2018, 38, 212–225, doi:10.1055/s-0038-1649502.
107. Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreirós, N.; Henke, M.; et al. Lack of Ceramide Synthase 2 Suppresses the Development of Experimental Autoimmune Encephalomyelitis by Impairing the Migratory Capacity of Neutrophils. Brain, Behavior, and Immunity 2015, 46, 280–292, doi:10.1016/j.bbi.2015.02.010.
108. Eberle, M.; Ebel, P.; Mayer, C.A.; Barthelmes, J.; Tafferner, N.; Ferreiros, N.; Ulshöfer, T.; Henke, M.; Foerch, C.; de Bazo, A.M.; et al. Exacerbation of Experimental Autoimmune Encephalomyelitis in Ceramide Synthase 6 Knockout Mice Is Associated with Enhanced Activation/Migration of Neutrophils. Immunology and Cell Biology 2015, 93, 825–836, doi:10.1038/icb.2015.47.
109. Schiffmann, S.; Ferreiros, N.; Birod, K.; Eberle, M.; Schreiber, Y.; Pfeilschifter, W.; Ziemann, U.; Pierre, S.; Scholich, K.; Grösch, S.; et al. Ceramide Synthase 6 Plays a Critical Role in the Development of Experimental Autoimmune Encephalomyelitis. The Journal of Immunology 2012, 188, 5723–5733, doi:10.4049/jimmunol.1103109.
110. Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshöfer, T.; Ferreirós, N.; Parnham, M.J.; Geisslinger, G.; et al. The Relevance of Ceramides and Their Synthesizing Enzymes for Multiple Sclerosis. Clinical Science 2018, 132, 1963–1976, doi:10.1042/CS20180506.
111. Vidaurre, O.G.; Haines, J.D.; Katz Sand, I.; Adula, K.P.; Huynh, J.L.; Mcgraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal Fluid Ceramides from Patients with Multiple Sclerosis Impair Neuronal Bioenergetics. Brain 2014, 137, 2271–2286, doi:10.1093/brain/awu139.
112. Sashindranath, M.; Nandurkar, H.H. Endothelial Dysfunction in the Brain: Setting the Stage for Stroke and Other Cerebrovascular Complications of Covid-19. Stroke 2021, 52, 1895–1904.
113. Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Milaganur Mohan, H.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral Ischemia Induces the Aggregation of Proteins Linked to Neurodegenerative Diseases. Scientific Reports 2018, 8, doi:10.1038/s41598-018-21063-z.
114. Kuźma, E.; Lourida, I.; Moore, S.F.; Levine, D.A.; Ukoumunne, O.C.; Llewellyn, D.J. Stroke and Dementia Risk: A Systematic Review and Meta-Analysis. Alzheimer’s and Dementia 2018, 14, 1416–1426, doi:10.1016/j.jalz.2018.06.3061.
115. Dmitrieva, V.G.; Torshina, E. v.; Yuzhakov, V. v.; Povarova, O. v.; Skvortsova, V.I.; Limborska, S.A.; Dergunova, L. v. Expression of Sphingomyelin Synthase 1 Gene in Rat Brain Focal Ischemia. Brain Research 2008, 1188, 222–227, doi:10.1016/j.brainres.2007.10.056.
116. Yu, Z.F.; Nikolova-Karakashian, M.; Zhou, D.; Cheng, G.; Schuchman, E.H.; Mattson, M.P. Pivotal Role for Acidic Sphingomyelinase in Cerebral Ischemia-Induced Ceramide and Cytokine Production, and Neuronal Apoptosis. Journal of Molecular Neuroscience 2000, 15, 85–97, doi:10.1385/JMN:15:2:85.
117. Hagemann, N.; Mohamud Yusuf, A.; Martiny, C.; Zhang, X.; Kleinschnitz, C.; Gunzer, M.; Kolesnick, R.; Gulbins, E.; Hermann, D.M. Homozygous Smpd1 Deficiency Aggravates Brain Ischemia/ Reperfusion Injury by Mechanisms Involving Polymorphonuclear Neutrophils, Whereas Heterozygous Smpd1 Deficiency Protects against Mild Focal Cerebral Ischemia. Basic Research in Cardiology 2020, 115, 64, doi:10.1007/s00395-020-00823-x.
118. Chao, H.C.; Lee, T.H.; Chiang, C.S.; Yang, S.Y.; Kuo, C.H.; Tang, S.C. Sphingolipidomics Investigation of the Temporal Dynamics after Ischemic Brain Injury. Journal of Proteome Research 2019, 18, 3470–3478, doi:10.1021/acs.jproteome.9b00370.
119. Gui, Y. kun; Li, Q.; Liu, L.; Zeng, P.; Ren, R.F.; Guo, Z.F.; Wang, G.H.; Song, J.G.; Zhang, P. Plasma Levels of Ceramides Relate to Ischemic Stroke Risk and Clinical Severity. Brain Research Bulletin 2020, 158, 122–127, doi:10.1016/j.brainresbull.2020.03.009.
120. Lee, T.H.; Cheng, C.N.; Chao, H.C.; Lee, C.H.; Kuo, C.H.; Tang, S.C.; Jeng, J.S. Plasma Ceramides Are Associated with Outcomes in Acute Ischemic Stroke Patients. Journal of the Formosan Medical Association 2021, doi:10.1016/j.jfma.2021.01.006.
121. Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12.
122. Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochemical Pharmacology 2015, 96, 119–130, doi:10.1016/j.bcp.2015.05.007.
123. Casasampere, M.; Ordóñez, Y.F.; Casas, J.; Fabrias, G. Dihydroceramide Desaturase Inhibitors Induce Autophagy via Dihydroceramide-Dependent and Independent Mechanisms. Biochimica et Biophysica Acta - General Subjects 2017, 1861, 264–275, doi:10.1016/j.bbagen.2016.11.033.
124. Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy of Cancer Cells via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229, doi:10.1080/15548627.2016.1213927.
125. van Brooklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine Kinase-1 Expression Correlates with Poor Survival of Patients with Glioblastoma Multiforme: Roles of Sphingosine Kinase Isoforms in Growth of Glioblastoma Cell Lines. Journal of Neuropathology and Experimental Neurology 2005, 64, 695–705, doi:10.1097/01.jnen.0000175329.59092.2c.
126. Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting Sphingosine Kinase 1 Inhibits Akt Signaling, Induces Apoptosis, and Suppresses Growth of Human Glioblastoma Cells and Xenografts. Cancer Research 2009, 69, 6915–6923, doi:10.1158/0008-5472.CAN-09-0664.
127. Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The Rotenone Model of Parkinson’s Disease: Genes, Environment and Mitochondria. Parkinsonism and Related Disorders 2003, 9, 59–64, doi:10.1016/S1353-8020(03)00023-3.
128. Schneider, J.S. MPTP‐induced Parkinsonism: Acceleration of Biochemical and Behavioral Recovery by GM1 Ganglioside Treatment. Journal of Neuroscience Research 1992, 31, 112–119, doi:10.1002/jnr.490310116.
129. Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Current Neurology and Neuroscience Reports 2018, 18, doi:10.1007/s11910-018-0860-4.
130. Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L. Selective Loss of Glucocerebrosidase Activity in Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Molecular Neurodegeneration 2015, 10, 1–6, doi:10.1186/s13024-015-0010-2.
131. Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P. Cerebrospinal Fluid β-Glucocerebrosidase Activity Is Reduced in Parkinson’s Disease Patients. Movement Disorders 2009, 34, 1423–1431, doi:10.1002/mds.27136.
132. Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658, doi:10.1093/brain/awv179.
133. Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced Glucocerebrosidase Is Associated with Increased α-Synuclein in Sporadic Parkinson’s Disease. Brain 2014, 137, 834–848, doi:10.1093/brain/awt367.
134. Taguchi, Y. v; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. Journal of Neuroscience 2017, 37, 9617–9631, doi:10.1523/JNEUROSCI.1525-17.2017.
135. Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52, doi:10.1016/j.cell.2011.06.001.
136. Kim, M.J.; Jeon, S.; Burbulla, L.F.; Krainc, D. Acid Ceramidase Inhibition Ameliorates α-Synuclein Accumulation upon Loss of GBA1 Function. Human Molecular Genetics 2018, 27, 1972–1988, doi:10.1093/hmg/ddy105.
137. Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E. Glucosylceramide Synthase Inhibition Alleviates Aberrations in Synucleinopathy Models. Proceedings of the National Academy of Sciences of the United States of America 2017, 114, 2699–2704, doi:10.1073/pnas.1616152114.
138. Burbulla, L.F.; Jeon, S.; Zheng, J.; Song, P.; Silverman, R.B.; Krainc, D. A Modulator of Wild-Type Glucocerebrosidase Improves Pathogenic Phenotypes in Dopaminergic Neuronal Models of Parkinson’s Disease. Science Translational Medicine 2019, 11, doi:10.1126/scitranslmed.aau6870.
139. Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Molecular Therapy 2017, 25, 2727–2742, doi:10.1016/j.ymthe.2017.08.004.
140. Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase Gene Therapy Prevents α-Synucleinopathy of Midbrain Dopamine Neurons. Neurobiology of Disease 2015, 82, 495–503, doi:10.1016/j.nbd.2015.09.009.
141. Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA Deficiency Promotes SNCA/α-Synuclein Accumulation through Autophagic Inhibition by Inactivated PPP2A. Autophagy 2015, 11, 1803–1820, doi:10.1080/15548627.2015.1086055.
142. Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hanniui, Y.A.; et al. Direct Interaction between the Inhibitor 2 and Ceramide via Sphingolipid‐protein Binding Is Involved in the Regulation of Protein Phosphatase 2A Activity and Signaling . The FASEB Journal 2009, 23, 751–763, doi:10.1096/fj.08-120550.
143. Salinas, M.; López-Valdaliso, R.; Martín, D.; Alvarez, A.; Cuadrado, A. Inhibition of PKB/Akt1 by C2-Ceramide Involves Activation of Ceramide- Activated Protein Phosphatase in PC12 Cells. Molecular and Cellular Neurosciences 2000, 15, 156–169, doi:10.1006/mcne.1999.0813.
144. Cornell, T.T.; Hinkovska-Galcheva, V.; Sun, L.; Cai, Q.; Hershenson, M.B.; Vanway, S.; Shanley, T.P. Ceramide-Dependent PP2A Regulation of TNFα-Induced IL-8 Production in Respiratory Epithelial Cells. American Journal of Physiology - Lung Cellular and Molecular Physiology 2009, 296, L849, doi:10.1152/ajplung.90516.2008.
145. Lleó, A.; Núñez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Paños, D.; Colom-Cadena, M.; Gomez-Giro, G.; Muñoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; et al. Changes in Synaptic Proteins Precede Neurodegeneration Markers in Preclinical Alzheimer’s Disease Cerebrospinal Fluid*. Molecular and Cellular Proteomics 2019, 18, 546–560, doi:10.1074/mcp.RA118.001290.
146. Launey, T.; Endo, S.; Sakai, R.; Harano, J.; Ito, M. Protein Phosphatase 2A Inhibition Induces Cerebellar Long-Term Depression and Declustering of Synaptic AMPA Receptor. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 676–681, doi:10.1073/pnas.0302914101.
147. Gao, J.; Hu, X.D.; Yang, H.; Xia, H. Distinct Roles of Protein Phosphatase 1 Bound on Neurabin and Spinophilin and Its Regulation in AMPA Receptor Trafficking and LTD Induction. Molecular Neurobiology 2018, 55, 7179–7186, doi:10.1007/s12035-018-0886-2.
148. Tong, L.; Prieto, G.A.; Cotman, C.W. IL-1β Suppresses CLTP-Induced Surface Expression of GluA1 and Actin Polymerization via Ceramide-Mediated Src Activation. Journal of Neuroinflammation 2018, 15, doi:10.1186/s12974-018-1158-9.
149. Furukawa, K.; Mattson, M.P. The Transcription Factor NF-ΚB Mediates Increases in Calcium Currents and Decreases in NMDA- and AMPA/Kainate-Induced Currents Induced by Tumor Necrosis Factor-α in Hippocampal Neurons. Journal of Neurochemistry 1998, 70, 1876–1886, doi:10.1046/j.1471-4159.1998.70051876.x.
150. Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-Mediated Macroautophagy Involves Inhibition of Protein Kinase B and Up-Regulation of Beclin 1. Journal of Biological Chemistry 2004, 279, 18384–18391, doi:10.1074/jbc.M313561200.
151. Nguyen, A.P.T.; Tsika, E.; Kelly, K.; Levine, N.; Chen, X.; West, A.B.; Boularand, S.; Barneoud, P.; Moore, D.J. Dopaminergic Neurodegeneration Induced by Parkinson’s Disease-Linked G2019S LRRK2 Is Dependent on Kinase and GTPase Activity. Proceedings of the National Academy of Sciences of the United States of America 2020, 117, 17296–17307, doi:10.1073/pnas.1922184117.
152. Singh, A.; Zhi, L.; Zhang, H. LRRK2 and Mitochondria: Recent Advances and Current Views. Brain Research 2019, 1702, 96–104, doi:10.1016/j.brainres.2018.06.010.
153. Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson Disease: Challenges of Clinical Trials. Nature Reviews Neurology 2020, 16, 97–107, doi:110.1038/s41582-019-0301-2.
154. Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 Kinase Activity Alters Neuronal Autophagy by Disrupting the Axonal Transport of Autophagosomes. Current Biology 2021, 31, 2140-2154.e6, doi:10.1016/j.cub.2021.02.061.
155. Yahalom, G.; Greenbaum, L.; Israeli-Korn, S.; Fay-Karmon, T.; Livneh, V.; Ruskey, J.A.; Roncière, L.; Alam, A.; Gan-Or, Z.; Hassin-Baer, S. Carriers of Both GBA and LRRK2 Mutations, Compared to Carriers of Either, in Parkinson’s Disease: Risk Estimates and Genotype-Phenotype Correlations. Parkinsonism and Related Disorders 2019, 62, 179–184, doi:10.1016/j.parkreldis.2018.12.014.
156. Ortega, R.A.; Wang, C.; Raymond, D.; Bryant, N.; Scherzer, C.R.; Thaler, A.; Alcalay, R.N.; West, A.B.; Mirelman, A.; Kuras, Y.; et al. Association of Dual LRRK2 G2019S and GBA Variations with Parkinson Disease Progression. JAMA Network Open 2021, 4, 215845, doi:10.1001/jamanetworkopen.2021.5845.
157. Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 Deficiency Impacts Ceramide Metabolism in Brain. Biochemical and Biophysical Research Communications 2016, 478, 1141–1146, doi:10.1016/j.bbrc.2016.08.082.
158. Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 Kinase Activity Regulates Lysosomal Glucocerebrosidase in Neurons Derived from Parkinson’s Disease Patients. Nature Communications 2019, 10, 1–9, doi:10.1038/s41467-019-13413-w.
159. Weindel, C.G.; Bell, S.L.; Vail, K.J.; West, K.O.; Patrick, K.L.; Watson, R.O. LRRK2 Maintains Mitochondrial Homeostasis and Regulates Innate Immune Responses to Mycobacterium Tuberculosis. eLife 2020, 9, doi:10.7554/eLife.51071.
160. Saez-Atienzar, S.; Bonet-Ponce, L.; Blesa, J.R.; Romero, F.J.; Murphy, M.P.; Jordan, J.; Galindo, M.F. The LRRK2 Inhibitor GSK2578215A Induces Protective Autophagy in SH-SY5Y Cells: Involvement of Drp-1-Mediated Mitochondrial Fission and Mitochondrial-Derived ROS Signaling. Cell Death and Disease 2014, 5, e1368–e1368, doi:10.1038/cddis.2014.320.
161. Fugio, L.B.; Coeli-Lacchini, F.B.; Leopoldino, A.M. Sphingolipids and Mitochondrial Dynamic. Cells 2020, 9.
162. van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.W.; Plasterk, R.H.A.; Nollen, E.A.A. C. Elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation during Aging. PLoS Genetics 2008, 4, 1000027, doi:10.1371/journal.pgen.1000027.
163. Khateeb, S.; Flusser, H.; Ofir, R.; Shelef, I.; Narkis, G.; Vardi, G.; Shorer, Z.; Levy, R.; Galil, A.; Elbedour, K.; et al. PLA2G6 Mutation Underlies Infantile Neuroaxonal Dystrophy. American Journal of Human Genetics 2006, 79, 942–948, doi:10.1086/508572.
164. Tomiyama, H.; Yoshino, H.; Ogaki, K.; Li, L.; Yamashita, C.; Li, Y.; Funayama, M.; Sasaki, R.; Kokubo, Y.; Kuzuhara, S.; et al. PLA2G6 Variant in Parkinson’s Disease. Journal of Human Genetics 2011, 56, 401–403, doi:10.1038/jhg.2011.22.
165. Shen, T.; Hu, J.; Jiang, Y.; Zhao, S.; Lin, C.; Yin, X.; Yan, Y.; Pu, J.; Lai, H.Y.; Zhang, B. Early-Onset Parkinson’s Disease Caused by Pla2g6 Compound Heterozygous Mutation, a Case Report and Literature Review. Frontiers in Neurology 2019, 10, 915, doi:10.3389/fneur.2019.00915.
166. Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-Dependent Ca2+ Signalling Is a Novel Determinant of Parkinson’s Disease. Nature Communications 2016, 7, 1–14, doi:10.1038/ncomms10332.
167. Lin, G.; Lee, P.-T.; Chen, K.; Lin, W.-W.; Wang, L.; Bellen Correspondence, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to Alpha-Synuclein Gain. Cell Metabolism 2018, 28, 605–618, doi:10.1016/j.cmet.2018.05.019.
168. Follett, J.; Norwood, S.J.; Hamilton, N.A.; Mohan, M.; Kovtun, O.; Tay, S.; Zhe, Y.; Wood, S.A.; Mellick, G.D.; Silburn, P.A.; et al. The Vps35 D620N Mutation Linked to Parkinson’s Disease Disrupts the Cargo Sorting Function of Retromer. Traffic 2014, 15, 230–244, doi:10.1111/tra.12136.
169. Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N. Serum Lipid Alterations in GBA-Associated Parkinson’s Disease. Parkinsonism and Related Disorders 2017, 44, 58–65, doi:10.1016/j.parkreldis.2017.08.026.
170. Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson’s Disease. Molecular Neurodegeneration 2020, 15, 1–18, doi:10.1186/s13024-020-00367-7.
171. Kitagishi, Y.; Nakano, N.; Ogino, M.; Ichimura, M.; Minami, A.; Matsuda, S. PINK1 Signaling in Mitochondrial Homeostasis and in Aging (Review). International Journal of Molecular Medicine 2017, 39, 3–8, doi:10.3892/ijmm.2016.2827.
172. Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Molecular Cell 2009, 33, 627–638, doi:10.1016/j.molcel.2009.02.013.
173. Sawada, M.; Nakashima, S.; Kiyono, T.; Nakagawa, M.; Yamada, J.; Yamakawa, H.; Banno, Y.; Shinoda, J.; Nishimura, Y.; Nozawa, Y.; et al. P53 Regulates Ceramide Formation by Neutral Sphingomyelinase through Reactive Oxygen Species in Human Glioma Cells. Oncogene 2001, 20, 1368–78.
174. García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct Effect of Ceramide on the Mitochondrial Electron Transport Chain Leads to Generation of Reactive Oxygen Species: Role of Mitochondrial Glutathione. Journal of Biological Chemistry 1997, 272, 11369–11377, doi:10.1074/jbc.272.17.11369.
175. Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-Induced BOK Promotes Mitochondrial Fission in Preeclampsia. Cell Death and Disease 2018, 9, 1–18, doi:10.1038/s41419-018-0360-0.
176. Qi, Z.; Yang, W.; Liu, Y.; Cui, T.; Gao, H.; Duan, C.; Lu, L.; Zhao, C.; Zhao, H.; Yang, H. Loss of PINK1 Function Decreases PP2A Activity and Promotes Autophagy in Dopaminergic Cells and a Murine Model. Neurochemistry International 2011, 59, 572–581, doi:10.1016/j.neuint.2011.03.023.
177. Dobrowsky, R.T.; Kamibayashi, C.; Mumby, M.C.; Hannuns, Y.A. Ceramide Activates Heterotrimeric Protein Phosphatase 2A. 1993, 268, 15523–15530, doi:10.1016/S0021-9258(18)82288-8.
178. Malini, E.; Zampieri, S.; Deganuto, M.; Romanello, M.; Sechi, A.; Bembi, B.; Dardis, A. Role of LIMP-2 in the Intracellular Trafficking of β-Glucosidase in Different Human Cellular Models. FASEB Journal 2015, 29, 3839–3852, doi:10.1096/fj.15-271148.
179. Usenko, T.S.; Bezrukova, A.I.; Bogdanova, D.A.; Kopytova, A.E.; Senkevich, K.A.; Gracheva, E. v.; Timofeeva, A.A.; Miliukhina, I. v.; Zakharova, E.Y.; Emelyanov, A.K.; et al. Genetics Variants and Expression of the SCARB2 Gene in the Pathogenesis of Parkinson’s Disease in Russia. Neuroscience Letters 2021, 741, 135509, doi:10.1016/j.neulet.2020.135509.
180. Bras, J.; Guerreiro, R.; Darwent, L.; Parkkinen, L.; Ansorge, O.; Escott-Price, V.; Hernandez, D.G.; Nalls, M.A.; Clark, L.N.; Honig, L.S.; et al. Genetic Analysis Implicates APOE, SNCA and Suggests Lysosomal Dysfunction in the Etiology of Dementia with Lewy Bodies. Human molecular genetics 2014, 23, 6139–6146, doi:10.1093/hmg/ddu334.
181. Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genetics 2011, 7, doi:10.1371/journal.pgen.1002141.
182. Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s Disease Age at Onset Genome-Wide Association Study: Defining Heritability, Genetic Loci, and α-Synuclein Mechanisms. Movement Disorders 2019, 34, 866–875, doi:10.1002/mds.27659.
183. Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; Destefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nature Genetics 2014, 46, 989–993, doi:10.1038/ng.3043.
184. Matrone, C.; Dzamko, N.; Madsen, P.; Nyegaard, M.; Pohlmann, R.; Søndergaard, R. v.; Lassen, L.B.; Andresen, T.L.; Halliday, G.M.; Jensen, P.H.; et al. Mannose 6-Phosphate Receptor Is Reduced in -Synuclein Overexpressing Models of Parkinsons Disease. PLoS ONE 2016, 11, e0160501, doi:10.1371/journal.pone.0160501.
185. Rothaug, M.; Zunke, F.; Mazzulli, J.R.; Schweizer, M.; Altmeppen, H.; Lüllmann-Rauche, R.; Kallemeijn, W.W.; Gaspar, P.; Aerts, J.M.; Glatzel, M.; et al. LIMP-2 Expression Is Critical for β-Glucocerebrosidase Activity and α-Synuclein Clearance. Proceedings of the National Academy of Sciences of the United States of America 2014, 111, 15573–15578, doi:10.1073/pnas.1405700111.
186. Subramaniam, S.R.; Chesselet, M.F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Progress in Neurobiology 2013, 106–107, 17–32, doi:10.1016/j.pneurobio.2013.04.004.
187. Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinson’s Disease 2018, 2018, doi:10.1155/2018/9163040.
188. Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From Modelling to Implication in Parkinson’s Disease. Folia Neuropathologica 2019, 57, 317–326.
189. Cataldi, S.; Arcuri, C.; Hunot, S.; Légeron, F.-P.; Mecca, C.; Garcia-Gil, M.; Lazzarini, A.; Codini, M.; Beccari, T.; Tasegian, A.; et al. Neutral Sphingomyelinase Behaviour in Hippocampus Neuroinflammation of MPTP-Induced Mouse Model of Parkinson’s Disease and in Embryonic Hippocampal Cells. Mediators of Inflammation 2017, 1–8, doi:10.1155/2017/2470950.
190. Magrassi, L.; Adorni, L.; Montorfano, G.; Rapelli, S.; Butti, G.; Berra, B.; Milanesi, G. Vitamin D Metabolites Activate the Sphingomyelin Pathway and Induce Death of Glioblastoma Cells. Acta Neurochirurgica 1998, 140, 707–714, doi:10.1007/s007010050166.
191. Pyszko, J.A.; Strosznajder, J.B. The Key Role of Sphingosine Kinases in the Molecular Mechanism of Neuronal Cell Survival and Death in an Experimental Model of Parkinson’s Disease. Folia Neuropathologica 2014, 52, 260–269, doi:10.5114/fn.2014.45567.
192. Pyszko, J.; Strosznajder, J.B. Sphingosine Kinase 1 and Sphingosine-1-Phosphate in Oxidative Stress Evoked by 1-Methyl-4-Phenylpyridinium (MPP+) in Human Dopaminergic Neuronal Cells. Molecular Neurobiology 50, 38–48, doi:10.1007/s12035-013-8622-4.
193. Shao, Q. hang; Chen, Y.; Li, F. fang; Wang, S.; Zhang, X. ling; Yuan, Y. he; Chen, N. hong TLR4 Deficiency Has a Protective Effect in the MPTP/Probenecid Mouse Model of Parkinson’s Disease. Acta Pharmacologica Sinica 2019, 40, 1503–1512, doi:10.1038/s41401-019-0280-2.
194. Zhao, X.D.; Wang, F.X.; Cao, W.F.; Zhang, Y.H.; Li, Y. TLR4 Signaling Mediates AP-1 Activation in an MPTP-Induced Mouse Model of Parkinson’s Disease. International Immunopharmacology 2016, 32, 96–102, doi:10.1016/j.intimp.2016.01.010.
195. Mariucci, G.; Pagiotti, R.; Galli, F.; Romani, L.; Conte, C. The Potential Role of Toll-Like Receptor 4 in Mediating Dopaminergic Cell Loss and Alpha-Synuclein Expression in the Acute MPTP Mouse Model of Parkinson’s Disease. Journal of Molecular Neuroscience 2018, 64, 611–618, doi:10.1007/s12031-018-1057-7.
196. Conte, C.; Roscini, L.; Sardella, R.; Mariucci, G.; Scorzoni, S.; Beccari, T.; Corte, L. Toll Like Receptor 4 Affects the Cerebral Biochemical Changes Induced by MPTP Treatment. Neurochemical Research 2017, 42, 493–500, doi:10.1007/s11064-016-2095-6.
197. Kaur, G.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Kumar, A.; Arora, S.; Bungau, S. Role of TLR2 and TLR4 Signaling in Parkinson’s Disease: An Insight into Associated Therapeutic Potential. Journal of molecular neuroscience : MN 2021, doi:10.1007/s12031-021-01811-z.
198. Albi, E.; Cataldi, S.; Codini, M.; Mariucci, G.; Lazzarini, A.; Ceccarini, M.R.; Ferri, I.; Laurenti, M.E.; Arcuri, C.; Patria, F.; et al. Neutral Sphingomyelinase Increases and Delocalizes in the Absence of Toll-Like Receptor 4: A New Insight for MPTP Neurotoxicity. Prostaglandins and Other Lipid Mediators 2019, 142, 46–52, doi:10.1016/j.prostaglandins.2019.03.004.
199. Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The Novel Mechanism of Rotenone-Induced α-Synuclein Phosphorylation via Reduced Protein Phosphatase 2A Activity. International Journal of Biochemistry and Cell Biology 2016, 75, 34–44, doi:10.1016/j.biocel.2016.03.007.
200. Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of Apoptotic Erythrocyte Death by Rotenone. Toxicology 2012, 300, 132–137, doi:10.1016/j.tox.2012.06.007.
201. Rutkute, K.; Asmis, R.H.; Nikolova-Karakashian, M.N. Regulation of Neutral Sphingomyelinase-2 by GSH: A New Insight to the Role of Oxidative Stress in Aging-Associated Inflammation. Journal of Lipid Research 2007, 48, 2443–2452, doi:10.1194/jlr.M700227-JLR200.
202. Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chemical Reviews 2011, 111, 6387–6422, doi:10.1021/cr2002917.
203. Grammatikos, G.; Schoell, N.; Ferreirós, N.; Bon, D.; Herrmann, E.; Farnik, H.; Köberle, V.; Piiper, A.; Zeuzem, S.; Kronenberger, B.; et al. Serum Sphingolipidomic Analyses Reveal an Upregulation of C16-Ceramide and Sphingosine-1-Phosphate in Hepatocellular Carcinoma. Oncotarget 2016, 7, 18095–18105, doi:10.18632/oncotarget.7741.
204. Alberg, A.J.; Armeson, K.; Pierce, J.S.; Bielawski, J.; Bielawska, A.; Visvanathan, K.; Hill, E.G.; Ogretmen, B. Plasma Sphingolipids and Lung Cancer: A Population-Based, Nested Case-Control Study. Cancer Epidemiology Biomarkers and Prevention 2013, 22, 1374–1382, doi:10.1158/1055-9965.EPI-12-1424.
205. Govindarajah, N.; Clifford, R.; Bowden, D.; Sutton, P.A.; Parsons, J.L.; Vimalachandran, D. Sphingolipids and Acid Ceramidase as Therapeutic Targets in Cancer Therapy. Crit Rev Oncol Hematol 2019, 138, 104–111, doi:10.1016/j.critrevonc.2019.03.018.
206. Belarbi, K.; Cuvelier, E.; Bonte, M.A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.C. Glycosphingolipids and Neuroinflammation in Parkinson’s Disease. Molecular Neurodegeneration 2020, 15, 1–16.
207. Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered Ceramide Acyl Chain Length and Ceramide Synthase Gene Expression in Parkinson’s Disease. Movement Disorders 2014, 29, 518–526, doi:10.1002/mds.25729.
208. Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 2021, 9, 491, doi:10.3390/biomedicines9050491.
209. Xing, Y.; Tang, Y.; Zhao, L.; Wang, Q.; Qin, W.; Ji, X.; Zhang, J.; Jia, J. Associations between Plasma Ceramides and Cognitive and Neuropsychiatric Manifestations in Parkinson’s Disease Dementia. Journal of the Neurological Sciences 2016, 370, 82–87, doi:10.1016/j.jns.2016.09.028.
210. Mielke, M.M.; Maetzler, W.*; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Grä Ber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 9, 1–6, doi:10.1371/journal.pone.0073094.
211. Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M. Lipid Pathway Alterations in Parkinson’s Disease Primary Visual Cortex. PLoS ONE 2011, 6, 1–17, doi:10.1371/journal.pone.0017299.
212. Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1–10, doi:10.1177/1759091418781889.
213. Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. v No Evidence for Substrate Accumulation in Parkinson Brains with GBA Mutations. Movement Disorders 2015, 30, 1085–1089, doi:10.1002/mds.26278.
214. Stoessel, D.; Schulte, C.; Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Costa, A. Promising Metabolite Profiles in the Plasma and CSF of Early Clinical Parkinson’s Disease. Frontiers in Aging Neuroscience 2018, 10, 1–14, doi:10.3389/fnagi.2018.00051.
215. Boutin, M.; Sun, Y.; Shacka, J.J.; Auray-Blais, C. Tandem Mass Spectrometry Multiplex Analysis of Glucosylceramide and Galactosylceramide Isoforms in Brain Tissues at Different Stages of Parkinson Disease. Analytical Chemistry 2016, 88, 1856–1863, doi:10.1021/acs.analchem.5b04227.
216. Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented Frontal Cortex Diacylglycerol Levels in Parkinson’s Disease and Lewy Body Disease. PLoS ONE 2018, 13, 1–15, doi:10.1371/journal.pone.0191815.
217. Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, but Significant Downregulation of Active Complement C5 Fragment. Journal of Parkinson’s Disease 2021, 11, 221–232, doi:10.3233/JPD-202310.
218. Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice Lacking Major Brain Gangliosides Develop Parkinsonism. Neurochemical Research 2011, 36, 1706–1714, doi:10.1007/s11064-011-0437-y.
219. Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the Mechanism of Internalization of α-Synuclein into Microglia: Roles of Ganglioside GM1 and Lipid Raft. Journal of Neurochemistry 2009, 110, 400–411, doi:10.1111/j.1471-4159.2009.06150.x.
220. Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and Is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Scientific Reports 2019, 9, 1–12, doi:10.1038/s41598-019-42847-x.
221. Gaspar, R.; Pallbo, J.; Weininger, U.; Linse, S.; Sparr, E. Ganglioside Lipids Accelerate α-Synuclein Amyloid Formation. Biochimica et Biophysica Acta - Proteins and Proteomics 2018, 1866, 1062–1072, doi:10.1016/j.bbapap.2018.07.004.
222. Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of Ganglioside GM1 Correlates with Parkinson’s Disease in Mice and Humans. Journal of Neuroscience Research 2012, 90, 1997–2008, doi:10.1002/jnr.23090.
223. Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Medical Science Monitor 2017, 23, 5345–5353, doi:10.12659/MSM.904399.
224. Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 Plasma Concentration in Idiopathic Parkinson’s Disease: A Lipidomic Analysis. PLoS ONE 2017, 12, 1–13, doi:10.1371/journal.pone.0172348.
225. Xing, Y.; Tang, Y.; Zhao, L.; Wang, Q.; Qin, W.; Ji, X.; Zhang, J.; Jia, J. Associations between Plasma Ceramides and Cognitive and Neuropsychiatric Manifestations in Parkinson’s Disease Dementia. Journal of the Neurological Sciences 2016, 370, 82–87, doi:10.1016/j.jns.2016.09.028.
This entry is adapted from the peer-reviewed paper 10.3390/biom11070945