 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ernesto Salzano | + 2246 word(s) | 2246 | 2021-06-21 09:48:01 | | | |
| 2 | Vivi Li | Meta information modification | 2246 | 2021-06-30 04:35:28 | | |
Video Upload Options
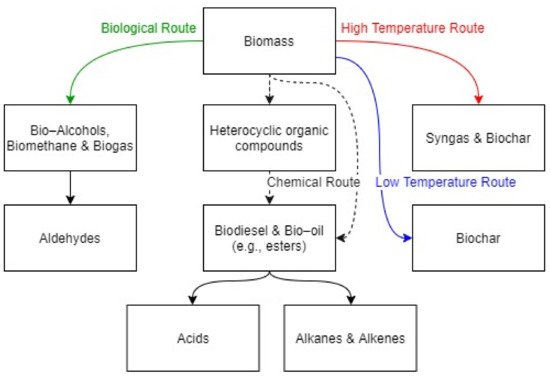
The need for lowering the environmental impacts has incentivized the investigation of biomass and biofuels as possible alternative sources for energy supply. Among the others, oxygenated bio-derived molecules such as alcohols, esters, acids, aldehydes, and furans are attractive substances as chemical feedstock and for sustainable energy production. Indeed, the presence of oxygen atoms limits the production of aromatic compounds, improves combustion efficiency (thus heat production) and alleviates the formation of carbon soot. On the other hand, the variability of their composition has represented one of the major challenges for the complete characterization of combustion behaviour. This work gives an overview of the current understanding of the detailed chemical mechanisms, as well as experimental investigations characterizing the combustion process of these species, with an emphasis on the laminar burning velocity and the ignition delay time.
1. Introduction
2. Combustion Chemistry of Oxy-Biofuels
2.1. Light Alcohols
2.2. Carboxylic Acids
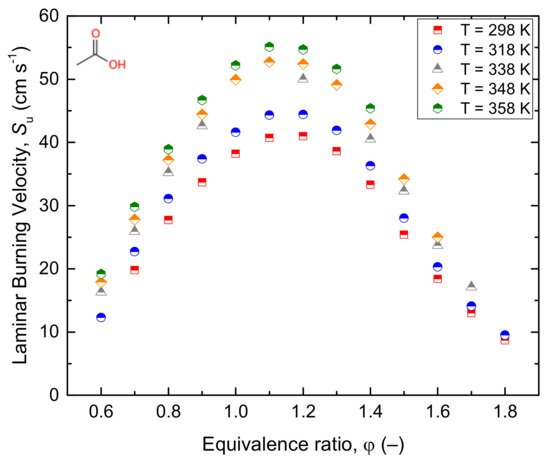
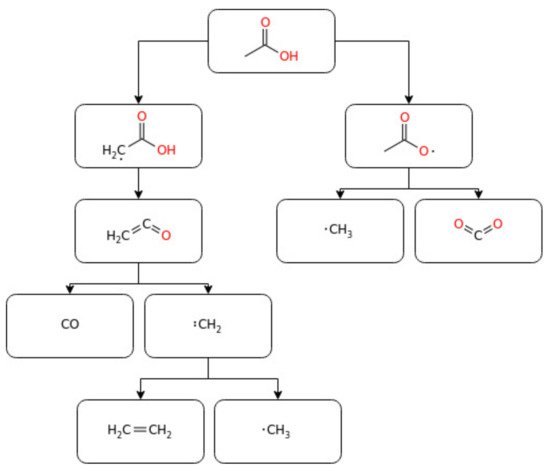
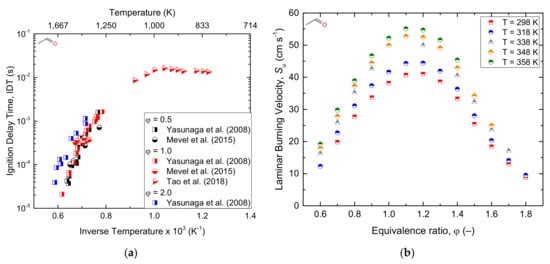
2.2.1. Acetic Acid
2.3. Light Aldehydes
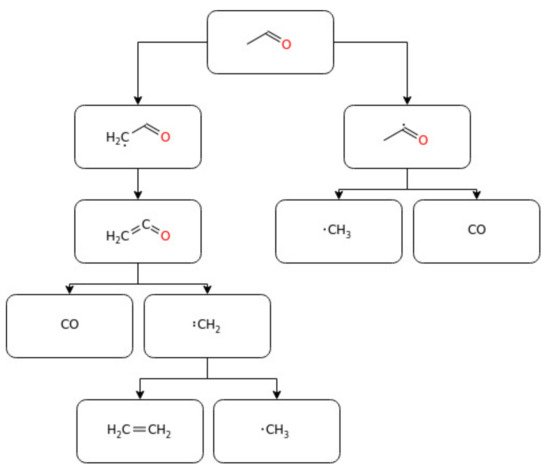
References
- Solomon, S.; Qin, D.; Manning, M.; Chen, Z.; Marquis, M.; Averyt, K.B.; Tignor, M.; Miller, H.L. Climate Change 2007—The Physical Science Basis: Working Group I Contribution to the Fourth Assessment Report of the IPCC; Cambridge University Press: Cambridge, MA, USA, 2007; Volume 4.
- Yasunaga, K.; Mikajiri, T.; Sarathy, S.M.; Koike, T.; Gillespie, F.; Nagy, T.; Simmie, J.M.; Curran, H.J. A shock tube and chemical kinetic modeling study of the pyrolysis and oxidation of butanols. Combust. Flame 2012, 159, 2009–2027.
- Agarwal, A.K. Biofuels (alcohols and biodiesel) applications as fuels for internal combustion engines. Prog. Energy Combust. Sci. 2007, 33, 233–271.
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634.
- Pelucchi, M.; Bissoli, M.; Rizzo, C.; Zhang, Y.; Somers, K.; Frassoldati, A.; Curran, H.; Faravelli, T. A Kinetic Modelling Study of Alcohols Operating Regimes in a HCCI Engine. SAE Int. J. Engines 2017, 10, 2354–2370.
- Namysl, S.; Pelucchi, M.; Herbinet, O.; Frassoldati, A.; Faravelli, T.; Battin-Leclerc, F. A first evaluation of butanoic and pentanoic acid oxidation kinetics. Chem. Eng. J. 2019, 373, 973–984.
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Battin-Leclerc, F.; et al. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part I: Reaction Classes, Rate Rules, Model Lumping, and Validation. Energy Fuels 2020, 34, 14688–14707.
- Veloo, P.S.; Wang, Y.L.; Egolfopoulos, F.; Westbrook, C.K. A comparative experimental and computational study of methanol, ethanol, and n-butanol flames. Combust. Flame 2010, 157, 1989–2004.
- Liu, D.; Togbé, C.; Tran, L.-S.; Felsmann, D.; Oßwald, P.; Nau, P.; Koppmann, J.; Lackner, A.; Glaude, P.-A.; Sirjean, B.; et al. Combustion chemistry and flame structure of furan group biofuels using molecular-beam mass spectrometry and gas chromatography—Part I: Furan. Combust. Flame 2014, 161, 748–765.
- Ubando, A.T.; Africa, A.D.M.; Maniquiz-Redillas, M.C.; Culaba, A.B.; Chen, W.-H. Reduction of particulate matter and volatile organic compounds in biorefineries: A state-of-the-art review. J. Hazard. Mater. 2021, 403, 123955.
- Nugroho, Y.K.; Zhu, L. Platforms planning and process optimization for biofuels supply chain. Renew. Energy 2019, 140, 563–579.
- Huheey, J.; Cottrell, T. The Strengths of Chemical Bonds; Butterworths: London, UK, 1958.
- Zhang, X.; Mei, B.; Ma, S.; Zhang, Y.; Cao, C.; Li, W.; Ye, L.; Li, Y. Characterizing the fuel-specific combustion chemistry of acetic acid and propanoic acid: Laminar flame propagation and kinetic modeling studies. Proc. Combust. Inst. 2020, 38, 449–457.
- Christensen, M.; Konnov, A.A. Laminar burning velocity of acetic acid + air flames. Combust. Flame 2016, 170, 12–29.
- Marshall, P.; Glarborg, P. Ab initio and kinetic modeling studies of formic acid oxidation. Proc. Combust. Inst. 2015, 35, 153–160.
- Li, Y.; Strathmann, T.J. Kinetics and mechanism for hydrothermal conversion of polyhydroxybutyrate (PHB) for wastewater valorization. Green Chem. 2019, 21, 5586–5597.
- Kohse-Höinghaus, K.; Osswald, P.; Cool, T.A.; Kasper, T.; Hansen, N.; Qi, F.; Westbrook, C.K.; Westmoreland, P.R. Biofuel Combustion Chemistry: From Ethanol to Biodiesel. Angew. Chem. Int. Ed. 2010, 49, 3572–3597.
- Saggese, C.; Frassoldati, A.; Cuoci, A.; Faravelli, T.; Ranzi, E. A wide range kinetic modeling study of pyrolysis and oxidation of benzene. Combust. Flame 2013, 160, 1168–1190.
- Cossu, R.; Liu, J.; Pivato, A.; Ragazzi, M. Biomass to biofuels: Challenges and opportunities. Renew. Energy 2020, 158, 1–2.
- Nau, P.; Seipel, A.; Lucassen, A.; Brockhinke, A.; Kohse-Höinghaus, K. Intermediate species detection in a morpholine flame: Contributions to fuel-bound nitrogen conversion from a model biofuel. Exp. Fluids 2010, 49, 761–773.
- Kong, J.; Liu, H.; Zheng, Z. Chemical Kinetics Study on Combustion of Ethanol/biodiesel/n-heptane. Renew. Energy 2020, 148, 150–167.
- Gainey, B.; Lawler, B. The role of alcohol biofuels in advanced combustion: An analysis. Fuel 2021, 283, 118915.
- Kwon, H.; Lapointe, S.; Zhang, K.; Wagnon, S.W.; Pitz, W.J.; Zhu, J.; McEnally, C.S.; Pfefferle, L.D.; Xuan, Y. Sooting tendencies of 20 bio-derived fuels for advanced spark-ignition engines. Fuel 2020, 276, 118059.
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Rodriguez, A.; Rizzo, C.; Somers, K.P.; Zhang, Y.; Herbinet, O.; Curran, H.J.; Faravelli, T. Combustion of n-C3–C6 Linear Alcohols: An Experimental and Kinetic Modeling Study. Part II: Speciation Measurements in a Jet-Stirred Reactor, Ignition Delay Time Measurements in a Rapid Compression Machine, Model Validation, and Kinetic Analysis. Energy Fuels 2020, 34, 14708–14725.
- Mack, J.H.; Schuler, D.; Butt, R.H.; Dibble, R. Experimental investigation of butanol isomer combustion in Homogeneous Charge Compression Ignition (HCCI) engines. Appl. Energy 2016, 165, 612–626.
- Svensson, E.; Tuner, M.; Verhelst, S. Influence of Injection Strategies on Engine Efficiency for a Methanol PPC Engine. SAE Tech. Pap. Ser. 2019, 2, 653–671.
- Togbé, C.; Halter, F.; Foucher, F.; Mounaim-Rousselle, C.; Dagaut, P. Experimental and detailed kinetic modeling study of 1-pentanol oxidation in a JSR and combustion in a bomb. Proc. Combust. Inst. 2011, 33, 367–374.
- Togbé, C.; Dagaut, P.; Mzé-Ahmed, A.; Diévart, P.; Halter, F.; Foucher, F. Experimental and Detailed Kinetic Modeling Study of 1-Hexanol Oxidation in a Pressurized Jet-Stirred Reactor and a Combustion Bomb. Energy Fuels 2010, 24, 5859–5875.
- Oasmaa, A.; Solantausta, Y.; Arpiainen, V.; Kuoppala, E.; Sipilä, K. Fast Pyrolysis Bio-Oils from Wood and Agricultural Residues. Energy Fuels 2010, 24, 1380–1388.
- Bertero, M.; de la Puente, G.; Sedran, U. Fuels from bio-oils: Bio-oil production from different residual sources, characterization and thermal conditioning. Fuel 2012, 95, 263–271.
- Onarheim, K.; Solantausta, Y.; Lehto, J. Process Simulation Development of Fast Pyrolysis of Wood Using Aspen Plus. Energy Fuels 2014, 29, 205–217.
- Doolan, K.R.; Mackie, J.; Reid, C.R. High temperature kinetics of the thermal decomposition of the lower alkanoic acids. Int. J. Chem. Kinet. 1986, 18, 575–596.
- Elwardany, A.; Nasir, E.F.; Es-Sebbar, E.; Farooq, A. Unimolecular decomposition of formic and acetic acids: A shock tube/laser absorption study. Proc. Combust. Inst. 2015, 35, 429–436.
- Zervas, E.; Montagne, X.; Lahaye, J. Influence of Fuel Composition on the Emission of Oxygenated Pollutants (Organic Acids, Alcohols and Carbonyl Compounds) from a SI Engine. Tech. Chron. Sci. J. 2004, 1, 49–58.
- Zervas, E.; Montagne, X.; Lahaye, J. Emission of Alcohols and Carbonyl Compounds from a Spark Ignition Engine. Influence of Fuel and Air/Fuel Equivalence Ratio. Environ. Sci. Technol. 2002, 36, 2414–2421.
- Leplat, N.; Vandooren, J. Numerical and experimental study of the combustion of acetic acid in laminar premixed flames. Combust. Flame 2012, 159, 493–499.
- Mackie, J.; Doolan, K.R. High-temperature kinetics of thermal decomposition of acetic acid and its products. Int. J. Chem. Kinet. 1984, 16, 525–541.
- Wagner, H.G.; Zabel, F. Zum thermischen Zerfall von Keten in der Gasphase. Ber. Bunsenges. Phys. Chem. 1971, 75, 114–118.
- Cavallotti, C.; Pelucchi, M.; Frassoldati, A. Analysis of acetic acid gas phase reactivity: Rate constant estimation and kinetic sim-ulations. Proc. Combust. Inst. 2019, 37, 539–546.
- Fontaras, G.; Karavalakis, G.; Kousoulidou, M.; Ntziachristos, L.; Bakeas, E.; Stournas, S.; Samaras, Z. Effects of low concentration biodiesel blends application on modern passenger cars. Part 2: Impact on carbonyl compound emissions. Environ. Pollut. 2010, 158, 2496–2503.
- Sarathy, S.M.; Oßwald, P.; Hansen, N.; Kohse-Höinghaus, K. Alcohol combustion chemistry. Prog. Energy Combust. Sci. 2014, 44, 40–102.
- Jacobson, M.Z. Effects of Ethanol (E85) versus Gasoline Vehicles on Cancer and Mortality in the United States. Environ. Sci. Technol. 2007, 41, 4150–4157.
- Gaffney, J.S.; Marley, N.A. The impacts of combustion emissions on air quality and climate—From coal to biofuels and beyond. Atmos. Environ. 2009, 43, 23–36.
- Yasunaga, K.; Kubo, S.; Hoshikawa, H.; Kamesawa, T.; Hidaka, Y. Shock-tube and modeling study of acetaldehyde pyrolysis and oxidation. Int. J. Chem. Kinet. 2008, 40, 73–102.
- Sivaramakrishnan, R.; Michael, J.V.; Harding, L.B.; Klippenstein, S. Resolving Some Paradoxes in the Thermal Decomposition Mechanism of Acetaldehyde. J. Phys. Chem. A 2015, 119, 7724–7733.
- Pelucchi, M.; Namysl, S.; Ranzi, E.; Frassoldati, A.; Herbinet, O.; Battin-Leclerc, F.; Faravelli, T. An experimental and kinetic modelling study of n-C4C6 aldehydes oxidation in a jet-stirred reactor. Proc. Combust. Inst. 2019, 37, 389–397.
- Mével, R.; Chatelain, K.; Catoire, L.; Green, W.H.; Shepherd, J.E. Chemical Kinetics of Acetaldehyde Pyrolysis and Oxidation. In Proceedings of the 9th US National Combustion Meeting, Cincinnati, OH, USA, 17–20 May 2015.
- Tao, T.; Sun, W.; Yang, B.; Hansen, N.; Moshammer, K.; Law, C.K. Investigation of the chemical structures of laminar premixed flames fueled by acetaldehyde. Proc. Combust. Inst. 2017, 36, 1287–1294.
- Christensen, M.; Abebe, M.T.; Nilsson, E.J.; Konnov, A.A. Kinetics of premixed acetaldehyde + air flames. Proc. Combust. Inst. 2015, 35, 499–506.
- Christensen, M.; Konnov, A.A. Laminar burning velocity of diacetyl + air flames. Further assessment of combustion chemistry of ketene. Combust. Flame 2017, 178, 97–110.
- Halstead, M.P.; Prothero, A.; Quinn, C.P. A mathematical model of the cool-flame oxidation of acetaldehyde. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1971, 322, 377–403.
- Felton, P.; Gray, B.; Shank, N. Low temperature oxidation in a stirred flow reactor—II. Acetaldehyde (theory). Combust. Flame 1976, 27, 363–376.
- Cavanagh, J.; Cox, R.; Olson, G. Computer modeling of cool flames and ignition of acetaldehyde. Combust. Flame 1990, 82, 15–39.
- Gibson, C.; Gray, P.; Griffiths, J.; Hasko, S. Spontaneous ignition of hydrocarbon and related fuels: A fundamental study of thermokinetic interactions. Symp. Int. Combust. 1985, 20, 101–109.
- Kaiser, E.W.; Westbrook, C.K.; Pitz, W.J. Acetaldehyde oxidation in the negative temperature coefficient regime: Experimental and modeling results. Int. J. Chem. Kinet. 1986, 18, 655–688.
- Pelucchi, M.; Ranzi, E.; Frassoldati, A.; Faravelli, T. Alkyl radicals rule the low temperature oxidation of long chain aldehydes. Proc. Combust. Inst. 2017, 36, 393–401.
- Pelucchi, M. Kinetic modeling of the low temperature cool flames of acetaldehyde in a well stirred reactor. In Proceedings of the Meeting of the Italian Section of the Combustion Institute, Lecce, Italy, 20–23 September 2015.
- Zhang, X.; Ye, L.; Li, Y.; Zhang, Y.; Cao, C.; Yang, J.; Zhongyue, Z.; Zhen, H.; Fei, Q. Acetaldehyde oxidation at low and intermediate temperatures: An experimental and kinetic modeling investigation. Combust. Flame 2018, 191, 431–441.
- Bentz, T.; Striebel, F.; Olzmann, M. Shock-Tube Study of the Thermal Decomposition of CH 3CHO and CH 3CHO + H Reaction. J. Phys. Chem. A 2008, 112, 6120–6124.
- Hidaka, Y.; Kubo, S.; Hoshikawa, T.; Wakamatsu, H. Shock-tube study of acetaldehyde pyrolysis. In Shock Waves; Springer: Berlin/Heidelberg, Germany, 2005; pp. 603–608.
- Ernst, J.; Spindler, K.; Wagner, H.G. Untersuchungen zum thermischen Zerfall von Acetaldehyd und Aceton. Ber. Bunsenges. Phys. Chem. 1976, 80, 645–650.


