 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Toshiyuki Matsunaga | + 2544 word(s) | 2544 | 2021-05-26 06:02:48 | | | |
| 2 | Nora Tang | Meta information modification | 2544 | 2021-06-24 05:37:25 | | |
Video Upload Options
AKR1B10 is a human nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reductase belonging to the aldo-keto reductase (AKR) 1B subfamily. It catalyzes the reduction of aldehydes, some ketones and quinones, and interacts with acetyl-CoA carboxylase and heat shock protein 90α. The enzyme is highly expressed in epithelial cells of the stomach and intestine, but down-regulated in gastrointestinal cancers and inflammatory bowel diseases. In contrast, AKR1B10 expression is low in other tissues, where the enzyme is upregulated in cancers, as well as in non-alcoholic fatty liver disease and several skin diseases. In addition, the enzyme’s expression is elevated in cancer cells resistant to clinical anti-cancer drugs. Thus, growing evidence supports AKR1B10 as a potential target for diagnosing and treating these diseases. Herein, we reviewed the literature on the roles of AKR1B10 in a healthy gastrointestinal tract, the development and progression of cancers and acquired chemoresistance, in addition to its gene regulation, functions, and inhibitors.
1. Introduction
Aldo-keto reductases (AKRs) are a group of NAD(P)(H)-dependent enzymes catalyzing interconversions between the carbonyl and alcohol groups of endogenous and xenobiotic compounds [1]. The AKR superfamily is systematized into 16 families: AKR1 (aldehyde reductases, aldose reductases, hydroxysteroid dehydrogenases, and steroid 5b-reductases); AKR2 (mannose and xylose reductases); AKR3 (yeast AKRs); AKR4 (chalcone and codeinone reductases); AKR5 (gluconic acid reductases); AKR6 (β-subunits of the potassiumgated voltage channels); AKR7 (aflatoxin dialdehyde and succinic semialdehyde reductases); AKR8 (pyridoxal reductases); AKR9 (aryl alcohol dehydrogenases); AKR10 (Streptomyces AKRs); AKR11 (Bacillus AKRs); AKR12 (Streptomyces sugar aldehyde reductases); AKR13 (hyperthermophilic bacteria reductases); AKR14 (Escherichia coli reductases), AKR15 (Mycobacterium reductases), and AKR16 (Vibrio cholerae reductases). Each family is further divided into several subfamilies based on a >60% amino acid sequence identity. To date, fifteen AKR members have been identified in humans and belong to the AKR1A, AKR1B, AKR1C, AKR1E, AKR6A, and AKR7A subfamilies. There are three members of the human AKR1B subfamily: AKR1B1 (aldose reductase), AKR1B10 (aldose reductase-like protein-1), and AKR1B15, whose genes are clustered at chromosome 7q33 [1]. AKR1B1, AKR1B10, and an enzymatically active isoform of AKR1B15 are 36-kDa soluble monomeric proteins consisting of 316 amino acids and sharing >68% amino acid sequence identity, of which 91.5% are shared between AKR1B10 and AKR1B15 [2][3]. The three AKRs are NADPH-dependent reductases and display overlapping substrate specificities for aromatic and aliphatic aldehydes but differ in their catalytic efficiencies [2][3][4][5][6], which is notably higher for retinal (all-trans-retinaldehyde) in AKR1B10 [5]. In addition, the glucose reductase activity characteristics of AKR1B1 are very low for AKR1B10 and AKR1B15 [2][4][5], and prostaglandin F synthase activity is observed with AKR1B1, but not with AKR1B10 [7]. In contrast to AKR1B1, AKR1B10 and AKR1B15 exhibit low 17β-hydroxysteroid dehydrogenase activity for estrone and 4-androstene-3,17-dione [3][6]. For subcellular localization, AKR1B1 and AKR1B10 are cytosolic, whereas AKR1B15 is in the mitochondria [3]. The three AKR1Bs also have different tissue distributions. While AKR1B1 is ubiquitous, AKR1B10 protein is predominantly expressed in the human stomach and intestine [2][8], although its mRNA is detected in many other tissues [3][8][9]. The mRNA for AKR1B15 is predominantly expressed in the placenta, testis, skeletal muscle, and adipose tissue, where its level is lower than that of mRNA for AKR1B10 [3].
2. Gene Regulation of AKR1B10
2.1. Factors Regulating AKR1B10 Expression

| Agent * | Signal Molecule | Cell ** | References |
|---|---|---|---|
| Up-regulation | |||
| Ethoxyquin | Nrf2 | Lung cancer A549, H23 | [14] |
| MG-132, bortezomib | Nrf2 | CRC SW-480, HT29 | [17] |
| Doxorubicin | Nrf2 | Gastric cancer MKN45 | [18] |
| EGF, insulin | AP-1, ERK | HCC HepG2, Hep3B | [19] |
| Lipopolysaccharide | Blood mononuclear cells | [20] | |
| BMP, IBMX | Mesenchymal stem cells | [21] | |
| 9,10-Phenanthrenequinone | Nrf2, ERK | Lung cancer A549 | [22] |
| Cigarette smoke extract | Airway epithelium | [23] | |
| Carnosic acid, t-BHQ | Nrf2 | Astrocytoma U373MG | [15] |
| 5-FU, L-OHP | p53 | CRC HT116 | [16] |
| Down-regulation | |||
| TPA | c-Jun, ERK | Lung cancer A549 | [15] |
| 5-FU, L-OHP | CRC HT29 | [16] |
2.2. Contribution of Nrf2 to AKR1B10 Induction
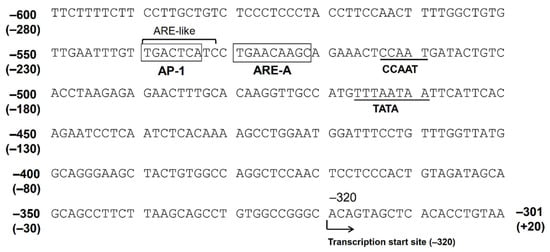
2.3. The Function of AP-1 Protein in AKR1B10 Gene Regulation
2.4. Signal Transduction
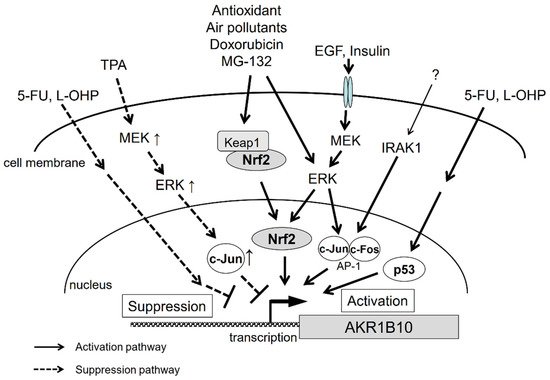
3. Conclusions
References
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 2015, 234, 236–246.
- Cao, D.; Fan, S.T.; Chung, S.S. Identification and characterization of a novel human aldose reductase-like gene. J. Biol. Chem. 1998, 273, 11429–11435.
- Weber, S.; Salabei, J.K.; Moller, G.; Kremmer, E.; Bhatnagar, A.; Adamski, J.; Barski, O.A. Aldo-keto Reductase 1B15 (AKR1B15): A mitochondrial human aldo-keto reductase with activity toward steroids and 3-keto-acyl-CoA conjugates. J. Biol. Chem. 2015, 290, 6531–6545.
- Endo, S.; Matsunaga, T.; Mamiya, H.; Ohta, C.; Soda, M.; Kitade, Y.; Tajima, K.; Zhao, H.T.; El-Kabbani, O.; Hara, A. Kinetic studies of AKR1B10, human aldose reductase-like protein: Endogenous substrates and inhibition by steroids. Arch. Biochem. Biophys. 2009, 487, 1–9.
- Gimenez-Dejoz, J.; Kolar, M.H.; Ruiz, F.X.; Crespo, I.; Cousido-Siah, A.; Podjarny, A.; Barski, O.A.; Fanfrlik, J.; Pares, X.; Farres, J.; et al. Substrate Specificity, Inhibitor Selectivity and Structure-Function Relationships of Aldo-Keto Reductase 1B15: A Novel Human Retinaldehyde Reductase. PLoS ONE 2015, 10, e0134506.
- Gimenez-Dejoz, J.; Weber, S.; Fernandez-Pardo, A.; Moller, G.; Adamski, J.; Porte, S.; Pares, X.; Farres, J. Engineering aldo-keto reductase 1B10 to mimic the distinct 1B15 topology and specificity towards inhibitors and substrates, including retinoids and steroids. Chem. Biol. Interact. 2019, 307, 186–194.
- Kabututu, Z.; Manin, M.; Pointud, J.C.; Maruyama, T.; Nagata, N.; Lambert, S.; Lefrancois-Martinez, A.M.; Martinez, A.; Urade, Y. Prostaglandin F2 Synthase Activities of Aldo-Keto Reductase 1B1, 1B3 and 1B7. J. Biochem. 2009, 145, 161–168.
- Fukumoto, S.; Yamauchi, N.; Moriguchi, H.; Hippo, Y.; Watanabe, A.; Shibahara, J.; Taniguchi, H.; Ishikawa, S.; Ito, H.; Yamamoto, S.; et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin. Cancer Res. 2005, 11, 1776–1785.
- Hyndman, D.J.; Flynn, T.G. Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim. Biophys. Acta 1998, 1399, 198–202.
- Oates, P.J. Aldose reductase, still a compelling target for diabetic neuropathy. Curr. Drug Targets 2008, 9, 14–36.
- Chang, K.C.; Petrash, J.M. Aldo-Keto Reductases: Multifunctional Proteins as Therapeutic Targets in Diabetes and Inflammatory Disease. Adv. Exp. Med. Biol. 2018, 1032, 173–202.
- Scuric, Z.; Stain, S.C.; Anderson, W.F.; Hwang, J.J. New member of aldose reductase family proteins overexpressed in human hepatocellular carcinoma. Hepatology 1998, 27, 943–950.
- Liu, Z.; Zhong, L.; Krishack, P.A.; Robbins, S.; Cao, J.X.; Zhao, Y.; Chung, S.; Cao, D. Structure and promoter characterization of aldo-keto reductase family 1 B10 gene. Gene 2009, 437, 39–44.
- Nishinaka, T.; Miura, T.; Okumura, M.; Nakao, F.; Nakamura, H.; Terada, T. Regulation of aldo-keto reductase AKR1B10 gene expression: Involvement of transcription factor Nrf2. Chem. Biol. Interact. 2011, 191, 185–191.
- Nishinaka, T.; Miura, T.; Sakou, M.; Hidaka, C.; Sasaoka, C.; Okamura, A.; Okamoto, A.; Terada, T. Down-regulation of aldo-keto reductase AKR1B10 gene expression by a phorbol ester via the ERK/c-Jun signaling pathway. Chem. Biol. Interact. 2015, 234, 274–281.
- Zinovieva, O.L.; Grineva, E.N.; Krasnov, G.S.; Karpov, D.S.; Zheltukhin, A.O.; Snezhkina, A.V.; Kudryavtseva, A.V.; Mashkova, T.D.; Lisitsyn, N.A. Treatment of cancer cells with chemotherapeutic drugs results in profound changes in expression of genes encoding aldehyde-metabolizing enzymes. J. Cancer 2019, 10, 4256–4263.
- Ebert, B.; Kisiela, M.; Wsol, V.; Maser, E. Proteasome inhibitors MG-132 and bortezomib induce AKR1C1, AKR1C3, AKR1B1, and AKR1B10 in human colon cancer cell lines SW-480 and HT-29. Chem. Biol. Interact. 2011, 191, 239–249.
- Morikawa, Y.; Kezuka, C.; Endo, S.; Ikari, A.; Soda, M.; Yamamura, K.; Toyooka, N.; El-Kabbani, O.; Hara, A.; Matsunaga, T. Acquisition of doxorubicin resistance facilitates migrating and invasive potentials of gastric cancer MKN45 cells through up-regulating aldo-keto reductase 1B10. Chem. Biol. Interact. 2015, 230, 30–39.
- Liu, Z.; Yan, R.; Al-Salman, A.; Shen, Y.; Bu, Y.; Ma, J.; Luo, D.X.; Huang, C.; Jiang, Y.; Wilber, A.; et al. Epidermal growth factor induces tumour marker AKR1B10 expression through activator protein-1 signalling in hepatocellular carcinoma cells. Biochem. J. 2012, 442, 273–282.
- Shaw, N.; Yang, B.; Millward, A.; Demaine, A.; Hodgkinson, A. AKR1B10 is induced by hyperglycaemia and lipopolysaccharide in patients with diabetic nephropathy. Cell Stress Chaperones 2014, 19, 281–287.
- Van Zoelen, E.J.; Duarte, I.; Hendriks, J.M.; van der Woning, S.P. TGFbeta-induced switch from adipogenic to osteogenic differentiation of human mesenchymal stem cells: Identification of drug targets for prevention of fat cell differentiation. Stem Cell Res. Ther. 2016, 7, 123.
- Matsunaga, T.; Morikawa, Y.; Haga, M.; Endo, S.; Soda, M.; Yamamura, K.; El-Kabbani, O.; Tajima, K.; Ikari, A.; Hara, A. Exposure to 9,10-phenanthrenequinone accelerates malignant progression of lung cancer cells through up-regulation of aldo-keto reductase 1B10. Toxicol. Appl. Pharmacol. 2014, 278, 180–189.
- Wang, R.; Wang, G.; Ricard, M.J.; Ferris, B.; Strulovici-Barel, Y.; Salit, J.; Hackett, N.R.; Gudas, L.J.; Crystal, R.G. Smoking-induced upregulation of AKR1B10 expression in the airway epithelium of healthy individuals. Chest 2010, 138, 1402–1410.
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193.
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639.
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139.
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521.
- Mimura, J.; Inose-Maruyama, A.; Taniuchi, S.; Kosaka, K.; Yoshida, H.; Yamazaki, H.; Kasai, S.; Harada, N.; Kaufman, R.J.; Oyadomari, S.; et al. Concomitant Nrf2- and ATF4-activation by Carnosic Acid Cooperatively Induces Expression of Cytoprotective Genes. Int. J. Mol. Sci. 2019, 20, 1706.
- MacLeod, A.K.; McMahon, M.; Plummer, S.M.; Higgins, L.G.; Penning, T.M.; Igarashi, K.; Hayes, J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: Demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009, 30, 1571–1580.
- Rooney, J.P.; Chorley, B.; Hiemstra, S.; Wink, S.; Wang, X.; Bell, D.A.; van de Water, B.; Corton, J.C. Mining a human transcriptome database for chemical modulators of NRF2. PLoS ONE 2020, 15, e0239367.
- Nishinaka, T.; Miura, T.; Shimizu, K.; Terada, T. Identification and characterization of functional antioxidant response elements in the promoter of the aldo-keto reductase AKR1B10 gene. Chem. Biol. Interact. 2017.
- Chiu, R.; Boyle, W.J.; Meek, J.; Smeal, T.; Hunter, T.; Karin, M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell 1988, 54, 541–552.
- Cheng, B.Y.; Lau, E.Y.; Leung, H.W.; Leung, C.O.; Ho, N.P.; Gurung, S.; Cheng, L.K.; Lin, C.H.; Lo, R.C.; Ma, S.; et al. IRAK1 Augments Cancer Stemness and Drug Resistance via the AP-1/AKR1B10 Signaling Cascade in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 2332–2342.
- Gimenez-Dejoz, J.; Weber, S.; Barski, O.A.; Moller, G.; Adamski, J.; Pares, X.; Porte, S.; Farres, J. Characterization of AKR1B16, a novel mouse aldo-keto reductase. Chem. Biol. Interact. 2017, 276, 182–193.
- Pastel, E.; Pointud, J.C.; Volat, F.; Martinez, A.; Lefrancois-Martinez, A.M. Aldo-Keto Reductases 1B in Endocrinology and Metabolism. Front. Pharmacol. 2012, 3, 148.


