Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanna D'Arcangelo | + 2972 word(s) | 2972 | 2021-06-21 05:49:05 | | | |
| 2 | Peter Tang | + 2 word(s) | 2974 | 2021-06-22 04:36:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
D'arcangelo, G. Neurodegeneration in NPC Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/11096 (accessed on 07 February 2026).
D'arcangelo G. Neurodegeneration in NPC Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/11096. Accessed February 07, 2026.
D'arcangelo, Giovanna. "Neurodegeneration in NPC Disease" Encyclopedia, https://encyclopedia.pub/entry/11096 (accessed February 07, 2026).
D'arcangelo, G. (2021, June 21). Neurodegeneration in NPC Disease. In Encyclopedia. https://encyclopedia.pub/entry/11096
D'arcangelo, Giovanna. "Neurodegeneration in NPC Disease." Encyclopedia. Web. 21 June, 2021.
Copy Citation
Niemann–Pick type C (NPC) disease is an autosomal recessive storage disorder, characterized by abnormal sequestration of unesterified cholesterol in the late endo-lysosomal system of cells. Progressive neurological deterioration and the onset of symptoms, such as ataxia, seizures, cognitive decline, and severe dementia, are pathognomonic features of the disease. In addition, different pathological similarities, including degeneration of hippocampal and cortical neurons, hyperphosphorylated tau, and neurofibrillary tangle formation, have been identified between NPC disease and other neurodegenerative pathologies.
Niemann–Pick type C
neurodegeneration
cognitive decline
lipid trafficking
pharmacological treatment
physical exercise
nutritional approach
1. Introduction
Niemann–Pick type C (NPC) disease is a rare, autosomal recessive neurovisceral disorder, caused in most cases by mutations in the NPC1 gene (95%), and only rarely in the NPC2 gene (5%) [1].
Lipid accumulation in the lysosomes and late endosomes is probably the crucial event in disease pathogenesis, although the underlying mechanisms are not fully understood [2]. Cholesterol has been widely recognized as the major storage lipid; however, other sphingolipid species might also be involved in the NPC pathogenesis [3][4]. Among these, sphingomyelin and glycosphingolipids have been characterized and well documented in both animal models and patients with NPC [5][6][7].
A further advance in understanding the NPC pathogenesis was the isolation of the two genes NPC1 and NPC2 and the subsequent elucidation of the role played by NPC proteins in cholesterol transport. Indeed, neurons are known to obtain cholesterol through endogenous synthesis or uptake of lipoprotein cholesterol particles produced and released within the central nervous system (CNS) [8]. Following internalization of these particles by target cells, unesterified cholesterol is transported from the endosomal/lysosomal system to the Golgi complex and endoplasmic reticulum, where it is processed and used as a substrate for further reactions [9] (Figure 1A). Brown and Goldstein suggested that the NPC1 and NPC2 proteins perform a combined activity during this process, as NPC2 binds unesterified cholesterol and transfers it to the N-terminal domain of membrane-associated NPC1, thereby allowing its transport out of the late endosome/lysosome compartment [10][11]. However, in the absence of NPC1 and NPC2, lipoprotein cholesterol particles remain trapped in late endosome/lysosome system, greatly reducing cholesterol levels in the Golgi complex and endoplasmic reticulum and causing deleterious effects on all those processes that depend on proper membrane composition [12] (Figure 1B).
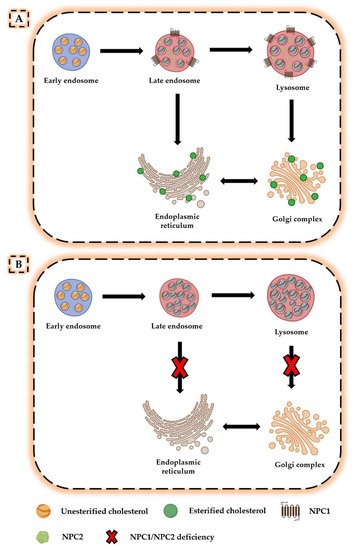
Figure 1. Altered cholesterol trafficking in Niemann–Pick disease type C. (A) In normal conditions, lipoprotein cholesterol particles bind to cell surface receptors and are internalized in the late endosome/lysosome system. In the presence of NPC1 and NPC2 proteins, unesterified cholesterol is transported from the late endosomal/lysosomal system to the Golgi complex and endoplasmic reticulum, where it is processed and used for other reactions. (B) In the absence of NPC1 or NPC2, unesterified cholesterol accumulates in the late endosomal/lysosomal system, resulting in deficiencies in the intracellular compartments for which it was intended.
The idea that NPC1 and NPC2 proteins act together is now generally accepted, as confirmed by subsequent studies in model systems and computationally [2][13][14]. It has also been proposed that NPC1 transfers cholesterol from the N-terminal domain to a sterol-sensing domain (SSD); however, the mechanism of this transfer is still unknown, in part because of the highly variable results agreeing only that NPC1 possesses a cholesterol-binding site aligned with the luminal leaflet of the lysosomal limiting membrane [15][16][17][18]. Mutations in the SSD domain or in the entire NPC1 protein can cause disease [19]. To understand how loss of NPC1 function leads to neurodegeneration typical of NPC disease, Reddy et al. using cDNA microarrays analyzed the genome-wide expression patterns of human fibroblasts homozygous for the I1061T NPC1 mutation, which is the most described and is characterized by a severe defect in intracellular processing of low-density lipoprotein (LDL)-derived cholesterol [20]. Homozygous carriers of the I1061T mutation manifest a relatively mild neurological form of the disease, with onset at a young age and homogeneous clinical symptoms. NPC1 fibroblasts showed highly significant differences from control cells, with an inappropriate homeostatic response to intracellular cholesterol accumulation. Indeed, it is known that LDL receptor expression is not down-regulated in NPC cells, so LDL uptake continues to occur despite increased cellular free cholesterol content [12][21]. Microarray analysis confirmed this dysregulation, as NPC fibroblasts homozygous for the I1061T mutation showed approximately 1.5-fold higher LDL receptor gene expression than normal fibroblasts [20]. In addition, the authors observed increased expression of oxysterol binding protein like 3 (OSBPL3), oxysterol binding protein like 6 (OSBPL6), and oxysterol binding protein like 8 (OSBPL8), which belong to the family of oxysterol-binding proteins involved in the non-vesicular cholesterol transfer between the endoplasmic reticulum and the plasma membrane [22]. OSBPL3 and OSBPL6 have been shown to contain sequences targeting for the endoplasmic reticulum and plasma membrane, and the subsequent overexpression of oxysterol binding protein 2 (OSBP2) promotes cholesterol synthesis and is responsible for LDL receptor upregulation, probably through enhancing endoplasmic reticulum cholesterol efflux [22]. Finally, NPC fibroblasts showed a gene expression profile indicative of oxidative stress, suggesting that all these changes may contribute to the pathophysiology of NPC disease [20]. However, among various unresolved questions, it remains to be clarified whether NPC1 protein is sufficient to complete cholesterol export or whether other entities are also involved.
Using a pharmacologically induced model (U18666a, a sterol molecule that potentially interferes with NPC1 protein function) of NPC disease in macrophages, Lloyd-Evans and colleagues studied the sequence of pathological events that occur in cells following NPC1 protein inactivation [23]. The authors found that NPC1 inactivation is followed by sphingosine accumulation and an approximately 75% reduction in lysosomal calcium levels [24]. The alteration in calcium homeostasis was attributed to sphingosine storage, as treatment of NPC1 cells with myriocin, which inhibits sphingolipid biosynthesis, normalized lysosomal calcium levels [24]. Notably, exogenous administration of sphingosine induced its rapid accumulation in lysosomes, confirming the role of sphingosines in rapidly translocating across membranes and its subsequent entrapment due to protonation [23][25]. These results suggest that the reduction in lysosomal calcium content and subsequent reduced calcium release from lysosomes is directly responsible for the endocytosis defects widely observed in NPC1 cells [24].
NPC disease has several nonspecific visceral, neurologic, and psychiatric clinical features that can arise at different stages of disease and progress at different rates [26]. Clinical symptoms are heterogeneous, with an age of onset ranging from the perinatal period to adulthood. The lifespan of patients also varies from a few days to over 60 years of age, although most cases die between 10 and 25 years of age [27][28][29]. Except for the few patients who die at birth or in the first 6 months of life from hepatic or respiratory failure, all will generally develop a progressive and fatal neurological disease, characterized by cerebellar ataxia, dysarthria, and dysphagia [30].
The disease severity and its onset might depend on the degree of functional disruption of cholesterol trafficking [31]. Particularly, an earlier onset of disease has been suggested in cases in which the degree of functional disruption of cholesterol trafficking is more severe, whereas a later onset and slower course might be associated with a less severe degree of functional disruption of cholesterol trafficking [29]. The disease severity is also related to a genetic component. In this regard, Wassif et al. analyzing NPC1 variants identified the p.H215R variant in almost one third of NPC1 alleles [32]. Furthermore, they demonstrated that two relatively common NPC1 variants with a combined carrier frequency approaching 0.8% may contribute, in compound heterozygous state, to a late-onset NPC1 phenotype for which the phenotypic spectrum and clinical significance have yet to be defined [32]. According to the authors, this late-onset NPC1 phenotype may represent a milder manifestation of NPC1 deficiency with predominantly visceral manifestations, although further studies are needed to define the degree to which this phenotype is associated with high frequency NPC1 alleles. Thus, the true prevalence of NPC disease is difficult to assess, due to insufficient clinical awareness, incomplete ascertainment of atypical phenotypes, as well as limitations of current diagnostic tests [32].
2. Neurodegeneration in NPC Disease
Given the key role of cholesterol in the regulation of membrane biophysical properties and cellular functions through the modulation of several signaling pathways, alterations in cholesterol homeostasis have been associated with disruption of brain function and the onset of neurodegeneration [33] (Figure 2). In this regard, Schultz et al. reported that the neuronal loss characterizing NPC disease is related not only to the composition and morphology of synaptic vesicles, but also to endosomal organelle transport [34]. Furthermore, Malnar and colleagues explained that the characteristic ataxia of NPC patients was closely dependent on the increased vulnerability of Purkinje cells in the cerebellum to the disease [35]. Npc1−/− mice, which are used in experimental practice as animal models for NPC1 disease, also mimic most of the pathologic features of NPC patients [36][37][38]. In fact, they exhibit not only loss of neurons, but also increased levels of cholesterol in mitochondria from brain and hepatocytes, highlighting the close relationship between the two organelles in cholesterol trafficking [34][39][40].
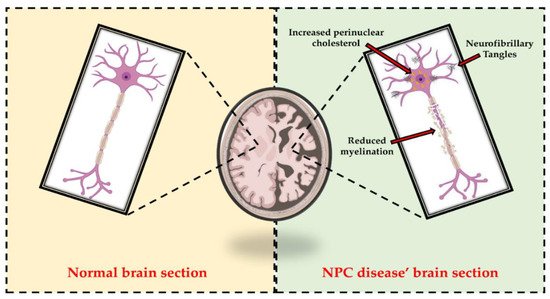
Figure 2. Representative comparison between normal brain section and NPC disease’ brain section. As highlighted in the right panel, alterations in cholesterol homeostasis are associated with disruption of brain function and the onset of neurodegeneration. In fact, increased perinuclear cholesterol, reduced myelination, and neurofibrillary tangles (NFT) accumulation are among the hallmarks of NPC patients’ neurons.
Notably, several pathological similarities have been identified between NPC and other neurodegenerative disorders, such as Alzheimer’s disease (AD). Particularly, progressive neurodegeneration, cholesterol accumulation and subsequent late endosome/lysosome abnormalities, Tau hyperphosphorylation, neurofibrillary tangles (NFTs), and β-Amyloid (Aβ) accumulation are known to occur in both NPC and AD [33][41][42]. In fact, although amyloid plaque formation has not been observed in the brains of NPC patients, in vivo and in vitro studies have shown that the altered cholesterol trafficking observed in NPC disease may modulate β-amyloid precursor protein (APP) processing [35]. Specifically, Malnar et al. observed in Npc1−/− cells a decreased APP expression on the cell surface and increased APP processing through the β-secretase pathway, resulting in increased levels of C99 and Aβ intracellular. The authors suggested that this effect was dependent on increased cholesterol levels, since cholesterol depletion not only reversed APP expression on the cell surface but also reduced Aβ and C99 levels in Npc1−/− cells with values comparable to those observed in control cells [43]. Therefore, these results confirmed the role played by cholesterol in APP metabolism, demonstrating the existence of a correlation between cholesterol homeostasis, APP metabolism, and AD pathogenesis.
In agreement, other scientific evidence reported increased levels of Aβ, Aβ 42, and β-C-terminal fragments (β-CTF) in experimental models of NPC disease [44][45][46]. Specifically, Burns et al. showed that aberrant intracellular cholesterol transport in Npc1 mutant mice was associated with both profoundly altered β-CTF levels, γ-secretase activity, and subcellular distribution of presenilin-1 (PS-1) and increased formation of Aβ 40 and Aβ 42 peptides [47]. Since late endosomes have been reported to act as a site for Aβ and PS-1 accumulation in NPC cells [44][45], the authors using a sucrose gradient to separate late and early endosomes observed in Npc1 mutant mice that, while PS-1 was present in the endoplasmic reticulum, and also accumulated in organelles with similar buoyancy to the early endosomal fractions (Rab 5 positive), no PS-1 was detected in the late endosomal fractions (Rab 7 positive). Furthermore, confocal microscopy analysis showed that PS-1 and Rab 5 immunoreactivity overlapped in brain tissue of Npc1 mutant mice but not in control mice. These findings, which agree with recent data from human NPC postmortem brains [46], highlight the role of cholesterol in amyloidogenesis, and provide information about the characterization of the Npc1 mutant mouse model that may help identify common pathological features between AD and NPC disease [47].
Gene expression analysis conducted by Reddy et al. of fibroblasts from patients homozygous for the I1061T NPC1 mutation revealed many interesting similarities with classic neurodegenerative diseases [20]. First, a significant increase in the generation of β-CTF at the protein level in human NPC1 fibroblasts compared with normal fibroblasts was confirmed. Genes encoding for LDL receptor-related proteins (LRP1, LRP2, and LRP6) were also upregulated by 1.8-, 2.2-, and 2.7-fold, respectively, in NPC fibroblasts. In addition, both mRNA and amyloid-beta precursor binding protein 2 (APBB2), which is known to interact with the cytoplasmic domain of APP and allow its cleavage by γ-secretase [48], were significantly upregulated by approximately 2.5-fold, thus contributing to the increased generation of Aβ 42 in NPC cells [49]. Based on their findings, the authors concluded that genes associated with AD are upregulated in NPC cells [20].
Subsequently, Kagedal and colleagues measured gene and protein expression of NPC1 in three distinct regions of the human brain, observing that its expression was upregulated in both the hippocampus and frontal cortex of AD patients compared with control subjects, whereas no difference was detected in the cerebellum [50]. In addition, a stronger expression of NPC1 was observed in hippocampal neurons, as well as reduced total cholesterol levels were found in the hippocampus of AD patients compared with control individuals. Thus, in agreement with other studies, it was suggested that the increased expression of NPC1 was related to altered cholesterol homeostasis in AD [50].
Recently, microglial changes have also been considered among the primary pathological events in neurodegeneration in NPC disease. Particularly, it has been observed that NPC1 plays a key role in the formation and maintenance of CNS myelin by oligodendrocytes, and that an alteration in intraneuronal lipid transport is closely related to reduced oligodendrocyte maturation and significant white matter hypomyelination [51][52]. These findings were largely confirmed by neuroimaging and brain magnetic resonance imaging (MRI) studies, which revealed diffuse axonal and myelinated gray and white matter changes in NPC disease, as well as volumetric changes at the level of the cerebellum, hippocampus, cortex, thalamus, and caudate nuclei [53][54]. Notably, a reduction in these changes and in the progression of volume loss was observed following treatment of NPC patients with miglustat [55].
3. Therapeutic Approaches Used to Counteract Cognitive Deficits
Despite the prevalence of cognitive impairment and its negative impact on functioning and quality of life, there is currently no cure for NPC disease. Research on possible disease-modifying therapies began in the 1950s and focused on studying the therapeutic effect of lipid-lowering agents, since unesterified cholesterol was originally considered the major metabolite underlying biochemical damage [56][57][58]. However, several experimental evidences showed that therapies aimed at cholesterol reduction were ineffective in counteracting cognitive impairment [59]. In this regard, Erickson et al. showed that treatment with nifedipine, a calcium channel blocker that induces cholesterol efflux, and probucol, an inhibitor cell surface cholesterol exporter ABCA1, reduced hepatic cholesterol levels in Npc1 mutant mice but had no effect on disabling neurological symptoms [60]. Similarly, Beheregaray and colleagues investigating the effect of clofibrate, a peroxisomal proliferating agent, on intracellular cholesterol accumulation in cultured fibroblasts from NPC patients, showed that it was not helpful in treating the disease, but on the contrary contributed to increased cholesterol levels in the cells of these individuals [61]. Thus, although treatment with lipid-lowering agents caused reductions in hepatic and plasma cholesterol levels in experimental mouse models and NPC patients, it had no impact on the neurological progression of the disease [59].
4. Additional Non-Pharmacologic Approaches for Neurodegeneration
4.1. Physical Exercise
In recent years, numerous researchers have supported the importance of exercise in the prevention and treatment of neurodegenerative diseases [62][63][64]. The central aim of such research has been to understand the mechanisms by which exercise is able to reduce and delay the onset of the symptomatology of neurological disorders, to identify the best protocols for patients [65]. In this regard, it is now generally accepted that regular physical activity promotes the release of myokines and metabolites into the circulation during muscle contraction [66]. These molecules can cross the blood–brain barrier at the level of brain capillaries and influence the functions of neurons and glial cells, thus modifying neurotransmission in different brain regions [65].
More importantly, physical activity is known to influence and improve cognitive processes, such as memory and learning, in animal models through its actions on the hippocampus [67]. For example, exercise, such as running, has been shown to increase neurogenesis in the dentate gyrus, as well as improve LTP and mnemonic function [68][69][70]. Similar results have also been found in human studies, where aerobic exercise has been shown to increase hippocampal volume and reduce age-related decline in mnemonic function [71][72].
Despite numerous studies over the years, specific exercises cannot yet be prescribed to maximize their positive effects on cognitive processes. This also depends on the fact that the levels of molecules released during muscle contraction change during and after exercise. In addition, it is still unclear how brain functioning may vary with the type, intensity, and timing of exercise.
Regular physical activity, when combined with current drug therapy, could be effective in reducing the progression and course of NPC disease.Particularly, specific short- and long-term training protocols, appropriately designed in terms of workload and recovery periods, could induce adaptive changes in cognition and brain plasticity.
4.2. Nutrition
Nutritional treatments may be considered another interventional approach to reduce symptoms and increase the lifespan, although there is very limited evidence on NPC disease and the few studies in the literature have mostly been conducted in experimental animal models.
The ketogenic diet has been used in patients suffering from inherited metabolic diseases (IMD) that present with seizure disorders [73], and has been proposed to NPC patients associated with miglustat administration to reduce seizure activity and ameliorate gastrointestinal side effects linked to the pharmacological therapy [74].
The ketosis induced through the ketogenic diet could restrict generation of reactive oxygen species and, thus, might prevent apoptosis. In this perspective, the use of antioxidants in the diet might be suggested to reduce subcellular stress in NPC patients. There is already scientific evidence indicating how the use of antioxidants in the NPC animal model can be beneficial. Vitamin E supplementation in fact delayed loss of weight, enhanced coordination and locomotor function, and increased the lifespan, improving the neurological symptoms in the Npc1 mutant mice [75]. This result points out how the use of this vitamin in the diet could be useful for the treatment of NPC patients. Furthermore, beneficial effect of nicotinamide is also reported in counteracting cognitive impairment and enhancing survival [76]. (Figure 3)
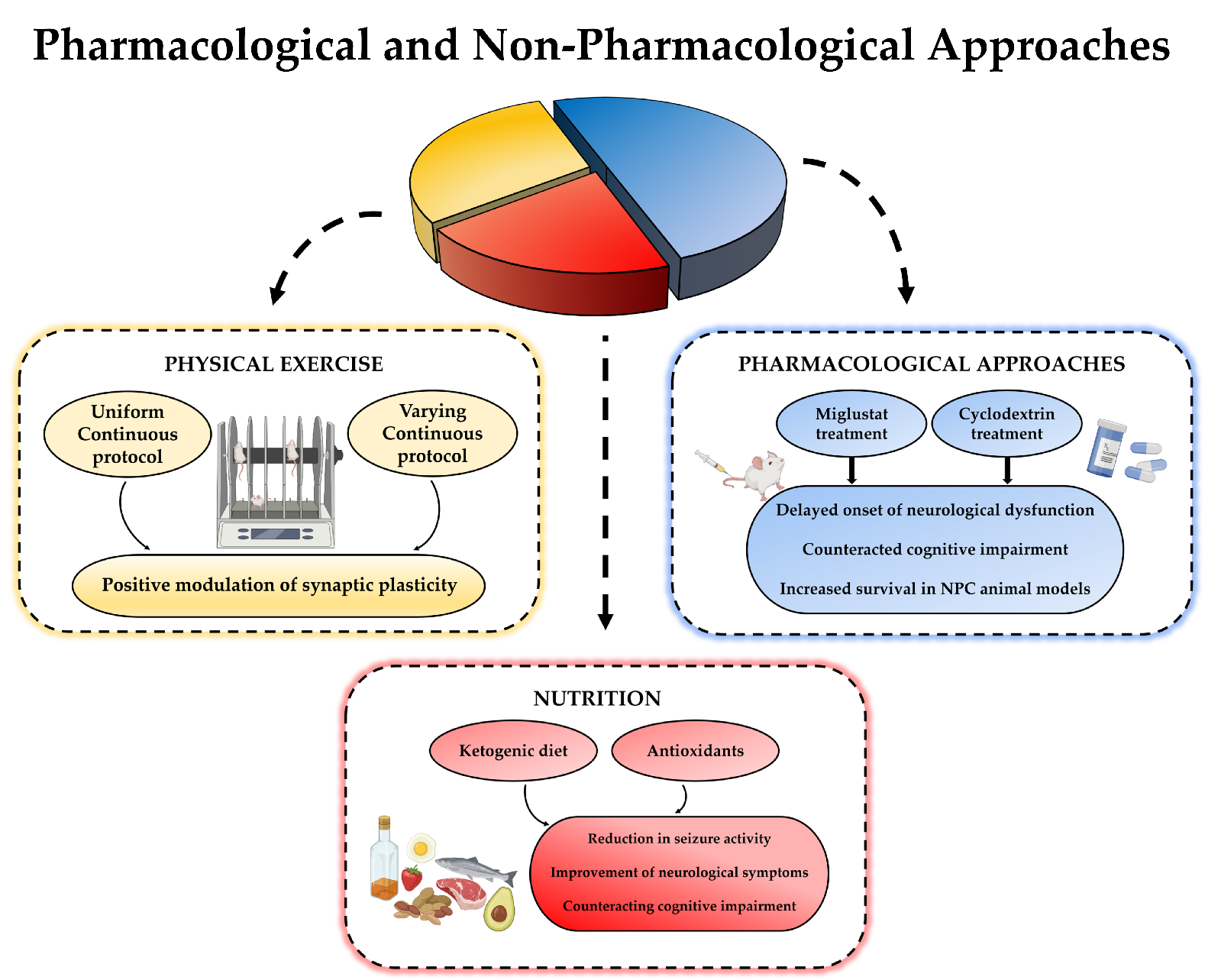
Figure 3. A schematic image of the combination between pharmacologic and non-pharmacologic approaches for NPC disease.
References
- Newton, J.; Milstien, S.; Spiegel, S. Niemann-Pick type C disease: The atypical sphingolipidosis. Adv. Biol. Regul. 2018, 70, 82–88.
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692.
- Bi, X.; Liao, G. Cholesterol in Niemann-Pick Type C Disease. In Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2010; Volume 51, pp. 319–335.
- Lloyd-Evans, E.; Platt, F.M. Lipids on trial: The search for the offending metabolite in Niemann-Pick type C disease. Traffic 2010, 11, 419–428.
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017, 31, 1719–1730.
- Scott, J.L.; Gabrielides, C.; Davidson, R.K.; Swingler, T.E.; Clark, I.M.; Wallis, G.A.; Boot-Handford, R.P.; Kirkwood, T.B.L.; Taylor, R.W.; Young, D.A. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 2010, 69, 1502–1510.
- Héron, B.; Valayannopoulos, V.; Baruteau, J.; Chabrol, B.; Ogier, H.; Latour, P.; Dobbelaere, D.; Eyer, D.; Labarthe, F.; Maurey, H.; et al. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2012, 7, 36.
- Mauch, D.H.; Nägler, K.; Schumacher, S.; Göritz, C.; Müller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357.
- Pagano, R.E. Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 885–891.
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010, 12, 166–173.
- Xie, X.; Brown, M.S.; Shelton, J.M.; Richardson, J.A.; Goldstein, J.L.; Liang, G. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15330–15335.
- Wojtanik, K.M.; Liscum, L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 2003, 278, 14850–14856.
- Berzina, Z.; Solanko, L.M.; Mehadi, A.S.; Jensen, M.L.V.; Lund, F.W.; Modzel, M.; Szomek, M.; Solanko, K.A.; Dupont, A.; Nielsen, G.K.; et al. Niemann-Pick C2 protein regulates sterol transport between plasma membrane and late endosomes in human fibroblasts. Chem. Phys. Lipids 2018, 213, 48–61.
- Hodošček, M.; Elghobashi-Meinhardt, N. Simulations of NPC1(NTD):NPC2 Protein Complex Reveal Cholesterol Transfer Pathways. Int. J. Mol. Sci. 2018, 19, 2623.
- Winkler, M.B.L.; Kidmose, R.T.; Szomek, M.; Thaysen, K.; Rawson, S.; Muench, S.P.; Wüstner, D.; Pedersen, B.P. Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 2019, 179, 485–497.e18.
- Elghobashi-Meinhardt, N. Computational Tools Unravel Putative Sterol Binding Sites in the Lysosomal NPC1 Protein. J. Chem. Inf. Model. 2019, 59, 2432–2441.
- Li, X.; Wang, J.; Coutavas, E.; Shi, H.; Hao, Q.; Blobel, G. Structure of human Niemann-Pick C1 protein. Proc. Natl. Acad. Sci. USA 2016, 113, 8212–8217.
- Wheeler, S.; Schmid, R.; Sillence, D.J. Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling. Int. J. Mol. Sci. 2019, 20, 717.
- Vanier, M.T.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281.
- Reddy, J.V.; Ganley, I.G.; Pfeffer, S.R. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS ONE 2006, 1, e19.
- Liscum, L.; Faust, J.R. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 1987, 262, 17002–17008.
- Olkkonen, V.M.; Levine, T.P. Oxysterol binding proteins: In more than one place at one time? Biochem. Cell Biol. 2004, 82, 87–98.
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255.
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca(2+) homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205.
- Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343.
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344.
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 2009, 149A, 446–450.
- Trendelenburg, G.; Vanier, M.T.; Maza, S.; Millat, G.; Bohner, G.; Munz, D.L.; Zschenderlein, R. Niemann-Pick type C disease in a 68-year-old patient. J. Neurol. Neurosurg. Psychiatry 2006, 77, 997–998.
- Sévin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2007, 130, 120–133.
- Garver, W.S.; Francis, G.A.; Jelinek, D.; Shepherd, G.; Flynn, J.; Castro, G.; Walsh Vockley, C.; Coppock, D.L.; Pettit, K.M.; Heidenreich, R.A.; et al. The National Niemann-Pick C1 disease database: Report of clinical features and health problems. Am. J. Med. Genet. A 2007, 143A, 1204–1211.
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16.
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016, 18, 41–48.
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382.
- Schultz, M.L.; Krus, K.L.; Lieberman, A.P. Lysosome and endoplasmic reticulum quality control pathways in Niemann-Pick type C disease. Brain Res. 2016, 1649, 181–188.
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72, 37–47.
- Elrick, M.J.; Pacheco, C.D.; Yu, T.; Dadgar, N.; Shakkottai, V.G.; Ware, C.; Paulson, H.L.; Lieberman, A.P. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 2010, 19, 837–847.
- Yu, T.; Shakkottai, V.G.; Chung, C.; Lieberman, A.P. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 2011, 20, 4440–4451.
- Lopez, M.E.; Klein, A.D.; Dimbil, U.J.; Scott, M.P. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 2011, 31, 4367–4378.
- Yu, W.; Gong, J.-S.; Ko, M.; Garver, W.S.; Yanagisawa, K.; Michikawa, M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005, 280, 11731–11739.
- Fernández, A.; Llacuna, L.; Fernández-Checa, J.C.; Colell, A. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 2009, 29, 6394–6405.
- Benussi, A.; Cotelli, M.S.; Padovani, A.; Borroni, B. Recent neuroimaging, neurophysiological, and neuropathological advances for the understanding of NPC. F1000Research 2018, 7, 194.
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann-Pick disease type C. Brain 1995, 118, 119–129.
- Malnar, M.; Kosicek, M.; Mitterreiter, S.; Omerbasic, D.; Lichtenthaler, S.F.; Goate, A.; Hecimovic, S. Niemann-Pick type C cells show cholesterol dependent decrease of APP expression at the cell surface and its increased processing through the beta-secretase pathway. Biochim. Biophys. Acta 2010, 1802, 682–691.
- Yamazaki, T.; Chang, T.Y.; Haass, C.; Ihara, Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J. Biol. Chem. 2001, 276, 4454–4460.
- Runz, H.; Rietdorf, J.; Tomic, I.; de Bernard, M.; Beyreuther, K.; Pepperkok, R.; Hartmann, T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J. Neurosci. 2002, 22, 1679–1689.
- Jin, L.-W.; Shie, F.-S.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 2004, 164, 975–985.
- Burns, M.; Gaynor, K.; Olm, V.; Mercken, M.; LaFrancois, J.; Wang, L.; Mathews, P.M.; Noble, W.; Matsuoka, Y.; Duff, K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 2003, 23, 5645–5649.
- Guénette, S.Y.; Chen, J.; Jondro, P.D.; Tanzi, R.E. Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10832–10837.
- Chang, Y.; Tesco, G.; Jeong, W.J.; Lindsley, L.; Eckman, E.A.; Eckman, C.B.; Tanzi, R.E.; Guénette, S.Y. Generation of the beta-amyloid peptide and the amyloid precursor protein C-terminal fragment gamma are potentiated by FE65L1. J. Biol. Chem. 2003, 278, 51100–51107.
- Kågedal, K.; Kim, W.S.; Appelqvist, H.; Chan, S.; Cheng, D.; Agholme, L.; Barnham, K.; McCann, H.; Halliday, G.; Garner, B. Increased expression of the lysosomal cholesterol transporter NPC1 in Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 831–838.
- Yu, T.; Lieberman, A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013, 9, e1003462.
- Takikita, S.; Fukuda, T.; Mohri, I.; Yagi, T.; Suzuki, K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J. Neuropathol. Exp. Neurol. 2004, 63, 660–673.
- Bowman, E.A.; Velakoulis, D.; Desmond, P.; Walterfang, M. Longitudinal Changes in White Matter Fractional Anisotropy in Adult-Onset Niemann-Pick Disease Type C Patients Treated with Miglustat. JIMD Rep. 2018, 39, 39–43.
- Masingue, M.; Adanyeguh, I.; Nadjar, Y.; Sedel, F.; Galanaud, D.; Mochel, F. Evolution of structural neuroimaging biomarkers in a series of adult patients with Niemann-Pick type C under treatment. Orphanet J. Rare Dis. 2017, 12, 22.
- Rego, T.; Farrand, S.; Goh, A.M.Y.; Eratne, D.; Kelso, W.; Mangelsdorf, S.; Velakoulis, D.; Walterfang, M. Psychiatric and Cognitive Symptoms Associated with Niemann-Pick Type C Disease: Neurobiology and Management. CNS Drugs 2019, 33, 125–142.
- Crocker, A.C.; Farber, S. Niemann-Pick disease: A review of eighteen patients. Medicine (Baltimore) 1958, 37, 1–95.
- Sylvain, M.; Arnold, D.L.; Scriver, C.R.; Schreiber, R.; Shevell, M.I. Magnetic resonance spectroscopy in Niemann-Pick disease type C: Correlation with diagnosis and clinical response to cholestyramine and lovastatin. Pediatr. Neurol. 1994, 10, 228–232.
- Patterson, M.C.; Di Bisceglie, A.M.; Higgins, J.J.; Abel, R.B.; Schiffmann, R.; Parker, C.C.; Argoff, C.E.; Grewal, R.P.; Yu, K.; Pentchev, P.G. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology 1993, 43, 61–64.
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140.
- Erickson, R.P.; Garver, W.S.; Camargo, F.; Hossain, G.S.; Heidenreich, R.A. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J. Inherit. Metab. Dis. 2000, 23, 54–62.
- Beheregaray, A.P.C.; Souza, F.T.S.; Coelho, J.C. Effect of a peroxysomal proliferator agent on intracellular cholesterol accumulation in cultured fibroblasts from Niemann-Pick type C disease patients. Clin. Chim. Acta 2003, 336, 137–142.
- Erickson, K.I.; Hillman, C.; Stillman, C.M.; Ballard, R.M.; Bloodgood, B.; Conroy, D.E.; Macko, R.; Marquez, D.X.; Petruzzello, S.J.; Powell, K.E. Physical Activity, Cognition, and Brain Outcomes: A Review of the 2018 Physical Activity Guidelines. Med. Sci. Sports Exerc. 2019, 51, 1242–1251.
- Hamer, M.; Chida, Y. Physical activity and risk of neurodegenerative disease: A systematic review of prospective evidence. Psychol. Med. 2009, 39, 3–11.
- Liu, Y.; Yan, T.; Chu, J.M.-T.; Chen, Y.; Dunnett, S.; Ho, Y.-S.; Wong, G.T.-C.; Chang, R.C.-C. The beneficial effects of physical exercise in the brain and related pathophysiological mechanisms in neurodegenerative diseases. Lab. Invest. 2019, 99, 943–957.
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes (Basel) 2019, 10, 720.
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392.
- Dahlin, E.; Andersson, M.; Thorén, A.; Hanse, E.; Seth, H. Effects of physical exercise and stress on hippocampal CA1 and dentate gyrus synaptic transmission and long-term potentiation in adolescent and adult Wistar rats. Neuroscience 2019, 408, 22–30.
- Brown, J.; Cooper-Kuhn, C.M.; Kempermann, G.; Van Praag, H.; Winkler, J.; Gage, F.H.; Kuhn, H.G. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur. J. Neurosci. 2003, 17, 2042–2046.
- Farmer, J.; Zhao, X.; van Praag, H.; Wodtke, K.; Gage, F.H.; Christie, B.R. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 2004, 124, 71–79.
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685.
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022.
- Firth, J.; Stubbs, B.; Vancampfort, D.; Schuch, F.; Lagopoulos, J.; Rosenbaum, S.; Ward, P.B. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage 2018, 166, 230–238.
- Scholl-Bürgi, S.; Höller, A.; Pichler, K.; Michel, M.; Haberlandt, E.; Karall, D. Ketogenic diets in patients with inherited metabolic disorders. J. Inherit. Metab. Dis. 2015, 38, 765–773.
- Höller, A.; Albrecht, U.; Baumgartner Sigl, S.; Zöggeler, T.; Ramoser, G.; Bernar, B.; Karall, D.; Scholl-Bürgi, S. Successful implementation of classical ketogenic dietary therapy in a patient with Niemann-Pick disease type C. Mol. Genet. Metab. Rep. 2021, 27, 100723.
- Marín, T.; Contreras, P.; Castro, J.F.; Chamorro, D.; Balboa, E.; Bosch-Morató, M.; Muñoz, F.J.; Alvarez, A.R.; Zanlungo, S. Vitamin E dietary supplementation improves neurological symptoms and decreases c-Abl/p73 activation in Niemann-Pick C mice. Nutrients 2014, 6, 3000–3017.
- Marshall, C.A.; Borbon, I.A.; Erickson, R.P. Relative efficacy of nicotinamide treatment of a mouse model of infantile Niemann-Pick C1 disease. J. Appl. Genet. 2017, 58, 99–102.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
22 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No